Selective vulnerability to kainate-induced oxidative damage in different rat brain regions
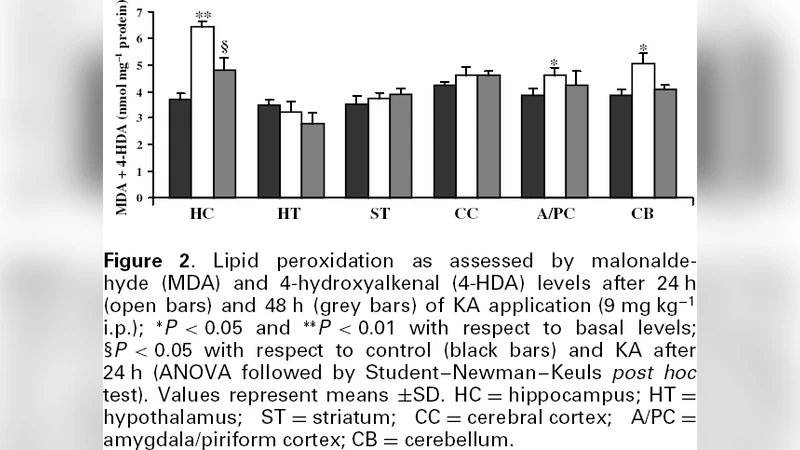
Some markers of oxidative injury were measured in different rat brain areas (hippocampus, cerebral cortex, striatum, hypothalamus, amygdala/piriform cortex and cerebellum) after the systemic administration of an excitotoxic dose of kainic acid (KA, 9 mg kg(-1) i.p.) at two different sampling times (24 and 48 h). Kainic acid was able to lower markedly (P < 0.05) the glutathione (GSH) levels in hippocampus, cerebellum and amygdala/piriform cortex (maximal reduction at 24 h). In a similar way, lipid peroxidation, as assessed by malonaldehyde and 4-hydroxyalkenal levels, significantly increased (P < 0.05) in hippocampus, cerebellum and amygdala/piriform cortex mainly at 24 h after KA. In addition, hippocampal superoxide dismutase (SOD) activity decreased significantly (P < 0.05) with respect to basal levels by 24 h after KA application. On the other hand, brain areas such as hypothalamus, striatum and cerebral cortex seem to be less susceptible to KA excitotoxicity. According to these findings, the pattern of oxidative injury induced by systemically administered KA seems to be highly region-specific. Further, our results have shown that a lower antioxidant status (GSH and SOD) seems not to play an important role in the selective vulnerability of certain brain regions because it correlates poorly with increases in markers of oxidative damage.
💡 Research Summary
The study investigated the regional specificity of oxidative injury induced by systemic administration of the excitotoxin kainic acid (KA) in rats. Adult rats received a single intraperitoneal injection of KA at a dose of 9 mg kg⁻¹, a concentration known to provoke seizures and excitotoxic damage. Brain tissue was harvested at two post‑injection time points, 24 hours and 48 hours, to capture both the acute peak of oxidative stress and any subsequent recovery or progression. Six anatomically and functionally distinct regions were examined: hippocampus, cerebral cortex, striatum, hypothalamus, amygdala/piriform cortex, and cerebellum. Within each region, three key biochemical indices were quantified: reduced glutathione (GSH) as a primary intracellular antioxidant, lipid peroxidation products (malondialdehyde, MDA, and 4‑hydroxy‑2‑nonenal, 4‑HNE) as markers of membrane oxidative damage, and superoxide dismutase (SOD) activity as a measure of enzymatic defense against superoxide radicals.
The results revealed a strikingly heterogeneous pattern of oxidative disruption. In the hippocampus, cerebellum, and amygdala/piriform cortex, GSH levels fell dramatically—by up to 50 % relative to controls—at the 24‑hour mark, indicating rapid depletion of the reduced thiol pool. Concomitantly, MDA and 4‑HNE concentrations rose significantly (p < 0.05), confirming extensive lipid peroxidation in these structures. Notably, hippocampal SOD activity was also reduced at 24 hours, suggesting that the enzymatic scavenging of superoxide was compromised specifically in this region. By 48 hours, GSH concentrations began to recover modestly, yet lipid peroxidation products remained elevated, implying that membrane damage persisted beyond the initial antioxidant collapse.
In contrast, the hypothalamus, striatum, and cerebral cortex displayed relative resilience. GSH content and SOD activity in these areas were either unchanged or showed only minor fluctuations, and lipid peroxidation markers did not reach statistical significance. This differential susceptibility aligns with known variations in kainate receptor density, mitochondrial oxidative capacity, and intrinsic antioxidant capacity across brain regions.
Importantly, the authors observed a weak correlation between the magnitude of antioxidant depletion (GSH, SOD) and the extent of oxidative damage (MDA, 4‑HNE). This finding challenges the simplistic view that lower baseline antioxidant status alone dictates vulnerability; instead, it points to a more complex interplay involving receptor-mediated calcium influx, mitochondrial dysfunction, and possibly region‑specific signaling cascades that amplify oxidative stress.
Methodologically, the study’s strength lies in its systematic, side‑by‑side comparison of multiple brain regions under identical systemic KA exposure, coupled with the use of two temporal checkpoints that capture both the acute oxidative surge and early recovery dynamics. However, limitations include the absence of histopathological correlation (e.g., neuronal loss, gliosis) and the lack of direct measurement of reactive oxygen species or downstream apoptotic markers, which would have enriched the mechanistic narrative.
Overall, the work demonstrates that systemic kainic acid elicits a highly region‑specific oxidative response in the rat brain. The hippocampus, cerebellum, and amygdala/piriform cortex emerge as the most vulnerable structures, showing pronounced glutathione depletion, lipid peroxidation, and, in the hippocampus, reduced SOD activity. Conversely, the hypothalamus, striatum, and cerebral cortex appear comparatively protected. The dissociation between antioxidant depletion and oxidative damage underscores that selective vulnerability cannot be explained solely by baseline antioxidant capacity; instead, it likely reflects differences in kainate receptor expression, calcium handling, and mitochondrial susceptibility. These insights have implications for designing targeted neuroprotective strategies that consider regional biochemical landscapes rather than relying on global antioxidant supplementation alone.
Comments & Academic Discussion
Loading comments...
Leave a Comment