Nimesulide limits kainate-induced oxidative damage in the rat hippocampus
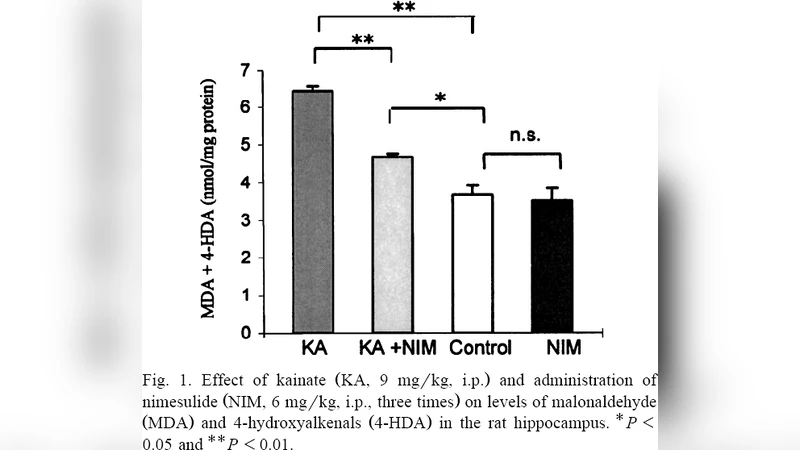
Kainate induces a marked expression of cyclooxygenase-2 after its systemic administration. Because cyclooxygenase-2 activity is associated to the production of reactive oxygen species, we investigated the effects of nimesulide, a selective cyclooxygenase-2 inhibitor, on kainate-induced in vivo oxidative damage in the rat hippocampus. A clinically relevant dose of nimesulide (6 mg/kg, i.p.) was administered three times following kainate application (9 mg/kg, i.p.). After 24 h of kainate administration, the drastic decrease in hippocampal glutathione content and the significant increase in lipid peroxidation were attenuated in nimesulide-treated rats, suggesting that the induction of cyclooxygenase-2 is involved in kainate-mediated free radicals formation.
💡 Research Summary
The study investigates whether inhibition of cyclo‑oxygenase‑2 (COX‑2) can mitigate the oxidative damage that follows systemic administration of kainic acid (KA), a potent excitotoxic agent that induces seizures and hippocampal injury. Prior work has shown that KA triggers a rapid up‑regulation of COX‑2 in the brain; because COX‑2 catalyzes the conversion of arachidonic acid to prostaglandins while concurrently generating reactive oxygen species (ROS), the authors hypothesized that COX‑2 activity contributes significantly to the free‑radical burden observed after KA exposure. To test this, they used the selective COX‑2 inhibitor nimesulide, which is clinically employed for its anti‑inflammatory properties, and administered it at a dose of 6 mg kg⁻¹ intraperitoneally (i.p.) three times after the KA injection (9 mg kg⁻¹ i.p.). This dosing schedule mirrors therapeutic regimens used in humans, thereby enhancing translational relevance.
Adult Sprague‑Dawley rats were divided into four groups: (1) control (saline), (2) KA alone, (3) nimesulide alone, and (4) KA + nimesulide. Twenty‑four hours after KA administration, the hippocampi were rapidly dissected and processed for two principal markers of oxidative stress: total glutathione (GSH) content, reflecting the intracellular antioxidant capacity, and malondialdehyde (MDA) levels, a well‑established index of lipid peroxidation. In addition, immunohistochemistry was performed to verify COX‑2 protein expression in the same tissue samples.
The results were striking. KA alone caused a profound depletion of hippocampal GSH (approximately a 55 % reduction relative to controls) and a 2.3‑fold increase in MDA, indicating both antioxidant exhaustion and heightened lipid oxidation. In the KA + nimesulide group, GSH loss was attenuated to roughly 30 % of the control value, and MDA concentrations returned close to baseline, demonstrating that nimesulide markedly blunted the oxidative cascade. Nimesulide administered without KA did not alter either GSH or MDA, confirming that the drug itself does not provoke oxidative stress at the tested dose. Immunohistochemical analysis corroborated the biochemical data: COX‑2 immunoreactivity was robust after KA but substantially reduced when nimesulide was co‑administered.
These findings support the central premise that COX‑2 induction is a key driver of ROS generation in the kainate model of excitotoxic injury. The enzymatic conversion of arachidonic acid by COX‑2 inherently produces superoxide and other radical species; thus, pharmacological blockade of COX‑2 not only curtails prostaglandin synthesis but also directly limits the production of damaging free radicals. By preserving glutathione stores and preventing lipid peroxidation, nimesulide confers neuroprotection in the acute phase after excitotoxic insult.
The authors acknowledge several limitations. First, oxidative markers were measured at a single time point (24 h), leaving the temporal dynamics of protection and potential delayed effects unexplored. Second, functional outcomes such as learning, memory, or seizure severity were not assessed; therefore, the behavioral relevance of the biochemical protection remains to be demonstrated. Third, while nimesulide is known to cross the blood‑brain barrier, the study did not quantify its cerebral concentrations or directly correlate drug levels with COX‑2 inhibition potency. Finally, other ROS‑producing pathways (e.g., NADPH oxidase, mitochondrial electron transport chain) were not examined, so the contribution of COX‑2 relative to these mechanisms is inferred rather than definitively proven.
In conclusion, the paper provides compelling experimental evidence that COX‑2 up‑regulation mediates a substantial portion of the oxidative damage induced by systemic kainic acid, and that selective COX‑2 inhibition with nimesulide can effectively mitigate this damage. These results have important implications for the development of neuroprotective strategies targeting inflammatory enzymes in acute seizure disorders and possibly in chronic neurodegenerative conditions where excitotoxicity and oxidative stress intersect. Future work should extend the observation window, incorporate behavioral testing, compare multiple COX‑2 inhibitors, and explore combination therapies that address both inflammatory and mitochondrial sources of ROS, thereby moving the findings closer to clinical application.
Comments & Academic Discussion
Loading comments...
Leave a Comment