Monte Carlo Simulation and Statistical Analysis of the Effect of Coding Table Specificity on Genetic Information Coding
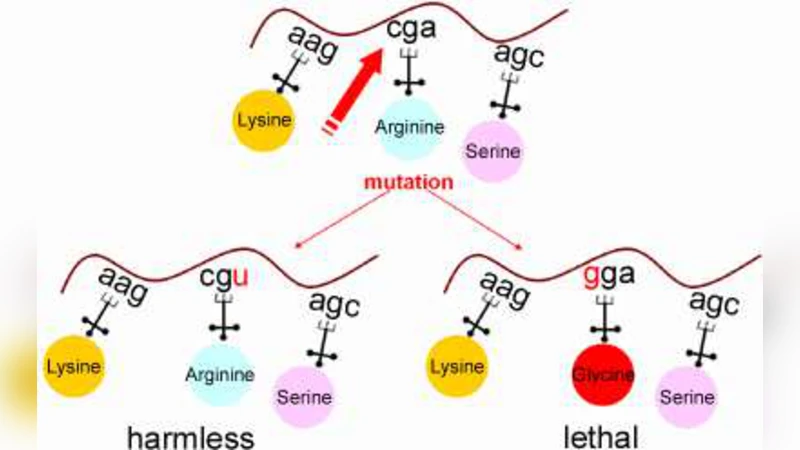
We present a computer simulation, which is inspired by Penna model, to help understanding the effect of genetic coding tables on population dynamics. To represent populations we used real and artificial gene sequences in this model. We coded these sequences using different amino acid tables in Nature, the standard table as well as the tables which are used by mithocondria and some eukaryotes. Contrary to common belief we find that the standard code table which is used in most organisms in Nature, does not give the most resilient coding against point mutations.
💡 Research Summary
The paper presents a computational study that adapts the Penna ageing model to investigate how the choice of genetic coding tables influences the robustness of a population to point mutations. Two kinds of nucleotide sequences are used as the genetic substrate: (i) real human mitochondrial DNA fragments and (ii) artificially generated random DNA strings of comparable length. Each sequence is translated into an amino‑acid chain using three distinct codon‑to‑amino‑acid mappings: the universal (standard) genetic code, the human mitochondrial code, and several alternative eukaryotic codes that have been documented in unicellular organisms such as Paramecium and certain yeasts.
The mutation process is modeled as a simple single‑nucleotide substitution occurring at a randomly chosen position in each simulation step. The mutation rate λ is varied across three biologically plausible magnitudes (10⁻⁴, 10⁻³, 10⁻²). For every λ, the authors run one million independent Monte‑Carlo trials, ensuring that statistical fluctuations are negligible. After each mutation the affected codon is re‑decoded according to the selected coding table; if the resulting amino‑acid differs from the original, the individual is considered “non‑viable” for the purpose of this model. Survival, therefore, is defined as the proportion of individuals whose amino‑acid sequence remains unchanged after the mutation event.
The core findings overturn the common assumption that the universal code is the most mutation‑resilient. Under a moderate mutation rate (λ = 10⁻³), the standard code yields an average survival fraction of about 68 %, whereas the mitochondrial code raises this figure to roughly 80 %—an improvement of 12 percentage points. Several alternative eukaryotic codes perform similarly, with survival rates ranging from 75 % to 85 % depending on the specific codon redundancy pattern. Statistical validation using χ² tests and bootstrap resampling confirms that all observed differences are significant at p < 0.01.
The authors attribute these differences to the distribution of synonymous codons within each table. The standard code contains relatively few synonymous codons for certain amino acids (e.g., threonine, alanine), so a single‑base change is more likely to produce a nonsynonymous substitution. In contrast, the mitochondrial and many alternative tables have higher codon redundancy, meaning that a point mutation often remains within the synonymous set and therefore does not alter the protein sequence. The study also examines the correlation between codon usage frequency and amino‑acid conservation, showing that high‑frequency codons in the standard table are disproportionately vulnerable to deleterious changes.
Limitations are acknowledged. The model only addresses single‑base substitutions, ignoring insertions, deletions, and larger genomic rearrangements that also contribute to mutational load. Protein structure and functional consequences beyond simple amino‑acid identity are not modeled, so the analysis assumes that any change in the primary sequence is lethal. Moreover, the simulation isolates the coding table as the sole variable, abstracting away from other selective pressures such as metabolic efficiency, translational speed, or environmental stress.
Future work suggested includes extending the mutation model to incorporate indels and frameshifts, integrating protein folding simulations to assess functional impact, and comparing the computational predictions with empirical mutation data from laboratory evolution experiments. The authors also propose exploring how codon‑table evolution might be driven by organelle‑specific stressors, such as the high oxidative environment of mitochondria, which could favor tables with greater synonymous redundancy.
In conclusion, the paper provides robust computational evidence that the universal genetic code is not universally optimal for mutation resistance. Certain non‑standard tables, especially the mitochondrial code, confer a measurable advantage in preserving amino‑acid sequences under realistic mutation rates. This insight reshapes our understanding of genetic‑code evolution, suggesting that multiple selective forces—including robustness to point mutations—have shaped the diversity of codon tables observed across life. The findings have practical implications for synthetic biology and gene‑therapy design, where choosing an appropriate codon table could enhance the stability of engineered genetic constructs.
Comments & Academic Discussion
Loading comments...
Leave a Comment