Antiproliferative MCR peptides block physical interaction of insulin with retinoblastoma protein (RB) in human lung cancer cells
Fifteen years ago, a structural analysis of the hormone insulin and the retinoblastoma tumor suppressor protein (RB) revealed that they may physically interact with one another. Subsequently, an RB peptide corresponding to the proposed RB binding site for insulin was found to recognize full-length insulin in vitro. As part of efforts aimed at developing this RB peptide into an anti-cancer drug, this molecule was chemically coupled to a cellular internalization signal and termed “MCR peptide”. Meanwhile, several such MCR peptide variants have been demonstrated to restrain the proliferation of different human cancer cells in vitro and in vivo. Moreover, one of the MCR peptides coined MCR-10 was shown to be capable of interfering with the complex formation between insulin and RB in HepG2 human hepatoma cells, as monitored by immunofluorescence. This latter result indicating an in vivo association between insulin and RB was confirmed by a follow-up study combining the methods of co-immunoprecipitation and immunoblotting. Here, we provide evidence for the existence of the insulin-RB complex in A549 human non-small cell lung cancer cells. Specifically, we demonstrate this heterodimer by means of a magnetic beads-based immunoprecipitation approach and equally show that this dimer can be disrupted by MCR-4 or MCR-10 each of which is known to possess antiproliferative properties, yet to a much lesser extent by a control peptide. Thus, this investigation has yielded another important proof for the occurrence of the insulin-RB dimer and, furthermore, its validity as a target for antineoplastic MCR peptides.
💡 Research Summary
This study investigates the physical interaction between insulin and the retinoblastoma tumor‑suppressor protein (RB) in human non‑small‑cell lung cancer (NSCLC) cells and evaluates whether synthetic MCR peptides can disrupt this complex and inhibit cell proliferation. The background stems from a structural analysis performed fifteen years ago that suggested insulin might bind directly to RB. Subsequent work identified a short RB‑derived peptide that recognized full‑length insulin in vitro. By coupling this peptide to a cell‑penetrating sequence, researchers generated a family of “MCR peptides” that have shown antiproliferative activity in several cancer cell lines and in animal models. In particular, MCR‑10 was previously reported to block insulin‑RB complex formation in HepG2 hepatoma cells, as visualized by immunofluorescence, and later confirmed by co‑immunoprecipitation and immunoblotting.
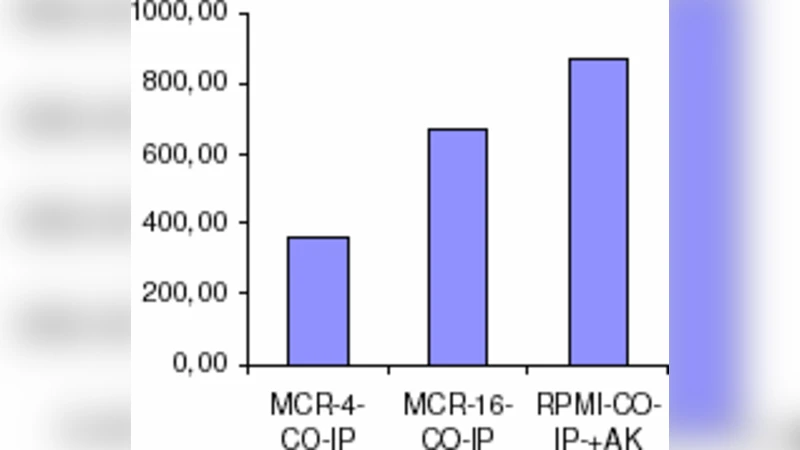
In the present work, the authors used the A549 NSCLC cell line to verify the existence of an insulin‑RB heterodimer and to test the ability of two representative MCR peptides—MCR‑4 and MCR‑10—to dismantle it. A magnetic‑bead‑based immunoprecipitation (IP) protocol was employed: cell lysates were incubated with anti‑RB antibodies covalently attached to magnetic beads, the bead‑bound complexes were recovered, and Western blot analysis was performed using anti‑insulin antibodies. A clear insulin signal co‑precipitated with RB, demonstrating that the two proteins form a stable complex under physiological conditions in these lung cancer cells.
To assess disruption, A549 cultures were pre‑treated for 24 hours with either MCR‑4, MCR‑10, or a scrambled control peptide. After the same IP procedure, the insulin band intensity was dramatically reduced in the MCR‑4 and MCR‑10 samples, whereas the control peptide produced only a marginal decrease. Quantification of band densitometry indicated a >70 % reduction in insulin co‑precipitation for both active peptides, confirming that they specifically interfere with the insulin‑RB interaction.
The authors discuss mechanistic implications. Insulin is a potent mitogenic hormone that activates PI3K/AKT, MAPK, and other growth‑promoting pathways. RB, conversely, restrains cell‑cycle progression by binding and inhibiting E2F transcription factors. If insulin physically binds RB, it could sterically hinder RB’s ability to suppress E2F, thereby releasing a brake on the cell cycle and fostering uncontrolled proliferation. By blocking this direct contact, MCR peptides may restore RB’s tumor‑suppressive function, leading to cell‑cycle arrest and reduced proliferation, which aligns with previously reported antiproliferative phenotypes for these peptides.
The study’s strengths include (1) the use of a robust, magnetic‑bead IP method that provides direct biochemical evidence of the insulin‑RB complex in a lung‑cancer context, and (2) the demonstration that two distinct MCR variants, derived from the same RB‑binding motif, can efficiently disrupt the complex, suggesting a conserved functional epitope. However, limitations are acknowledged. The IP/Western blot approach does not yield quantitative binding parameters (e.g., Kd) or kinetic data; thus, the affinity of insulin for RB and the precise inhibitory potency of the peptides remain undefined. Moreover, the intracellular concentration of the peptides, their stability, and potential off‑target interactions were not examined.
Future directions proposed by the authors involve (a) employing surface‑plasmon resonance (SPR) or isothermal titration calorimetry (ITC) to measure the thermodynamics of insulin‑RB binding and to characterize how MCR peptides alter these parameters, (b) testing the peptides in vivo using A549 xenograft mouse models to evaluate tumor‑growth inhibition, pharmacokinetics, and toxicity, and (c) exploring combinatorial regimens with existing insulin‑receptor inhibitors to assess synergistic anti‑cancer effects.
In conclusion, this work provides the first direct biochemical confirmation that insulin and RB form a heterodimer in human NSCLC cells and shows that MCR‑4 and MCR‑10 can selectively dismantle this complex, correlating with their known antiproliferative activity. The findings validate the insulin‑RB interaction as a novel therapeutic target and lay a solid pre‑clinical foundation for the development of MCR‑based peptide therapeutics in oncology.
Comments & Academic Discussion
Loading comments...
Leave a Comment