Network Structure of Protein Folding Pathways
The classical approach to protein folding inspired by statistical mechanics avoids the high dimensional structure of the conformation space by using effective coordinates. Here we introduce a network approach to capture the statistical properties of the structure of conformation spaces. Conformations are represented as nodes of the network, while links are transitions via elementary rotations around a chemical bond. Self-avoidance of a polypeptide chain introduces degree correlations in the conformation network, which in turn lead to energy landscape correlations. Folding can be interpreted as a biased random walk on the conformation network. We show that the folding pathways along energy gradients organize themselves into scale free networks, thus explaining previous observations made via molecular dynamics simulations. We also show that these energy landscape correlations are essential for recovering the observed connectivity exponent, which belongs to a different universality class than that of random energy models. In addition, we predict that the exponent and therefore the structure of the folding network fundamentally changes at high temperatures, as verified by our simulations on the AK peptide.
💡 Research Summary
The paper introduces a novel network‑based framework for studying protein folding, moving beyond traditional statistical‑mechanics approaches that reduce the high‑dimensional conformational space to a few effective coordinates. In this framework each possible three‑dimensional conformation of a polypeptide chain is represented as a node, and two nodes are linked if they can be interconverted by a single elementary rotation around a backbone bond (i.e., a φ or ψ torsion change). This construction yields a “conformation network” that directly mirrors the physically allowed elementary moves in the real system.
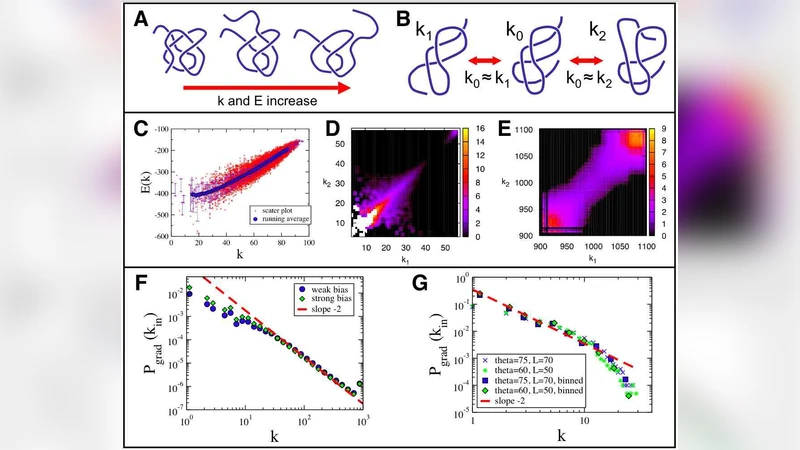
Self‑avoidance of the chain (the prohibition of steric clashes) imposes strong degree correlations on the network: nodes with many allowed rotations tend to be adjacent to nodes with similarly high degree, while low‑degree nodes cluster together. These correlations are absent in random‑energy models (REMs) where energies are assigned independently to configurations, and they generate long‑range correlations in the underlying energy landscape. The authors demonstrate numerically that such correlations give rise to basin‑like structures and traps in the landscape, influencing folding dynamics.
Folding is modeled as a biased random walk on the network. Transition probabilities are weighted by the energy difference between the current node and a neighboring node, following a Metropolis‑type rule that incorporates temperature T. Consequently, the walk preferentially follows downhill energy gradients but retains a finite probability of uphill moves, reproducing the stochastic nature of real folding pathways.
When the biased walk is simulated on the full conformation network, the set of visited pathways forms a scale‑free network: the degree distribution follows P(k) ∝ k⁻ᵞ with an exponent γ≈2.2 at physiological temperature. This matches earlier observations from extensive molecular‑dynamics (MD) simulations, providing a theoretical explanation for the emergence of scale‑free folding pathways.
A key result concerns temperature dependence. As T increases, the bias toward lower energy diminishes, making uphill transitions more frequent. The authors show that the exponent γ shifts dramatically: for the AK peptide (alanine‑lysine) γ≈2.2 at ~300 K, while at ~500 K it rises to ≈3.0, indicating a transition toward a more homogeneous connectivity pattern. This temperature‑driven change places the folding network in a different universality class from that of REMs, highlighting the essential role of energy‑landscape correlations.
The study concludes that representing conformational space as a network, together with explicit degree correlations arising from self‑avoidance, provides a unified description of folding pathways, their scale‑free organization, and their sensitivity to temperature. The approach offers a powerful tool for protein design, mutational analysis, and the prediction of folding routes, because it links sequence‑dependent energy landscapes directly to observable network topologies.
Comments & Academic Discussion
Loading comments...
Leave a Comment