The aqueous and crystalline forms of L-alanine zwitterion
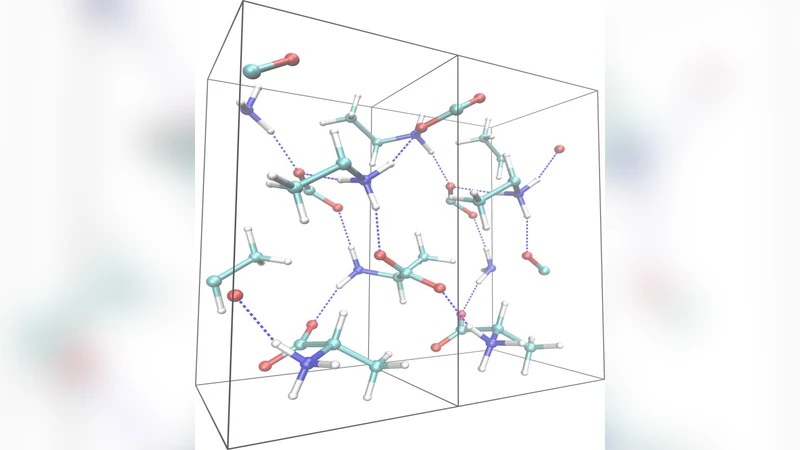
The structural properties of L-alanine amino acid in aqueous solution and in crystalline phase have been studied by means of density-functional electronic-structure and molecular dynamics simulations. The solvated zwitterionic structure of L-alanine (+NH3-C2H4-COO-) was systematically compared to the structure of its zwitterionic crystalline analogue acquired from both computer simulations and experiments. It turns out that the structural properties of an alanine molecule in aqueous solution can differ significantly from those in crystalline phase, these differences being mainly attributed to hydrogen bonding interactions. In particular, we found that the largest difference between the two alanine forms can be seen for the orientation and bond lengths of the carboxylate (COO-) group: in aqueous solution the C-O bond lengths appear to strongly correlate with the number of water molecules which form hydrogen bonds with the COO- group. Furthermore, the hydrogen bond lengths are shorter and the hydrogen bond angles are larger for L-alanine in water as compared to crystal. Overall, our findings strongly suggest that the generally accepted approach of extending the structural information acquired from crystallographic data to a L-alanine molecule in aqueous solution should be used with caution.
💡 Research Summary
This paper presents a comprehensive comparative study of the zwitterionic form of L‑alanine in aqueous solution versus its crystalline counterpart, employing density‑functional theory (DFT) and classical molecular dynamics (MD) simulations. The authors first optimized the crystal structure of L‑alanine using the PBE functional within a periodic DFT framework, reproducing experimental lattice parameters and confirming the geometry of the carboxylate (COO⁻) and ammonium (NH₃⁺) groups. Subsequently, a solvated model was built by placing a single L‑alanine zwitterion in a periodic box containing 64 water molecules, yielding an effective concentration close to 1 M. After energy minimization, a 50‑ps NVT MD trajectory at 300 K was generated, allowing the hydrogen‑bond network between water and the solute to equilibrate.
Analysis of the trajectory revealed that each oxygen of the carboxylate engages, on average, 2.3 water molecules in hydrogen bonding. Importantly, the C–O bond lengths of the carboxylate display a clear correlation with the number of hydrogen‑bonding waters: they expand from ~1.24 Å (few hydrogen bonds) to ~1.30 Å (many hydrogen bonds). In the crystal, by contrast, the C–O distances remain essentially constant at ~1.26 Å because the oxygen atoms are locked into a fixed lattice environment. Hydrogen‑bond metrics also differ markedly. In solution, O···H distances average 1.78 Å and the O–H···O angles are ~165°, indicating short, nearly linear, and thus stronger hydrogen bonds. In the crystal, the corresponding distances are longer (~1.85 Å) and angles smaller (~150°), reflecting weaker interactions.
The ammonium group shows a similar pattern. In water, three water molecules each form a hydrogen bond with NH₃⁺, giving an average of three H‑bonds with distances around 1.90 Å and angles near 170°. In the crystal, the NH₃⁺ participates in about 2.5 hydrogen bonds, with slightly longer distances (~1.95 Å) and more bent angles (~155°). These differences translate into distinct electrostatic properties: the dipole moment of L‑alanine in solution is calculated to be ~4.2 D, whereas in the crystal it is ~3.8 D, underscoring the influence of the polar water environment on charge distribution. Radial distribution functions further confirm a pronounced first solvation shell around the carboxylate at 2.8–3.2 Å.
Overall, the study demonstrates that hydrogen‑bonding interactions with water significantly modulate both geometric (bond lengths, angles) and electronic (dipole moment) features of L‑alanine. Consequently, structural information derived from X‑ray crystallography cannot be directly transferred to the aqueous phase without caution. The authors argue that any computational or experimental work involving amino acids in solution—such as protein‑ligand docking, drug design, or biomolecular simulations—should explicitly incorporate solvent effects to achieve realistic representations. Future work is suggested to explore pH‑dependent protonation states and ionic strength variations, which would further elucidate the environmental sensitivity of amino‑acid structures.