공유 유전적 제어 검정 방법 개선
본 논문은 GWAS에서 서로 다른 질환이나 형질 간에 동일한 인과 변이가 존재하는지를 검정하는 콜로컬라이제이션 테스트의 오류 원인을 규명하고, 변수 선택에 따른 타입Ⅰ 오류 상승을 방지하기 위해 주성분 분석과 베이지안 모델 평균화를 이용한 두 가지 새로운 접근법을 제안한다. Graves병과 Hashimoto 갑상선염 데이터에 적용한 결과, 13개 위험 영역 중 7곳에서 실제 공유 유전 신호가 확인되었으며, 기존 방법이 과도하게 허위 양성을 일으…
저자: Chris Wallace
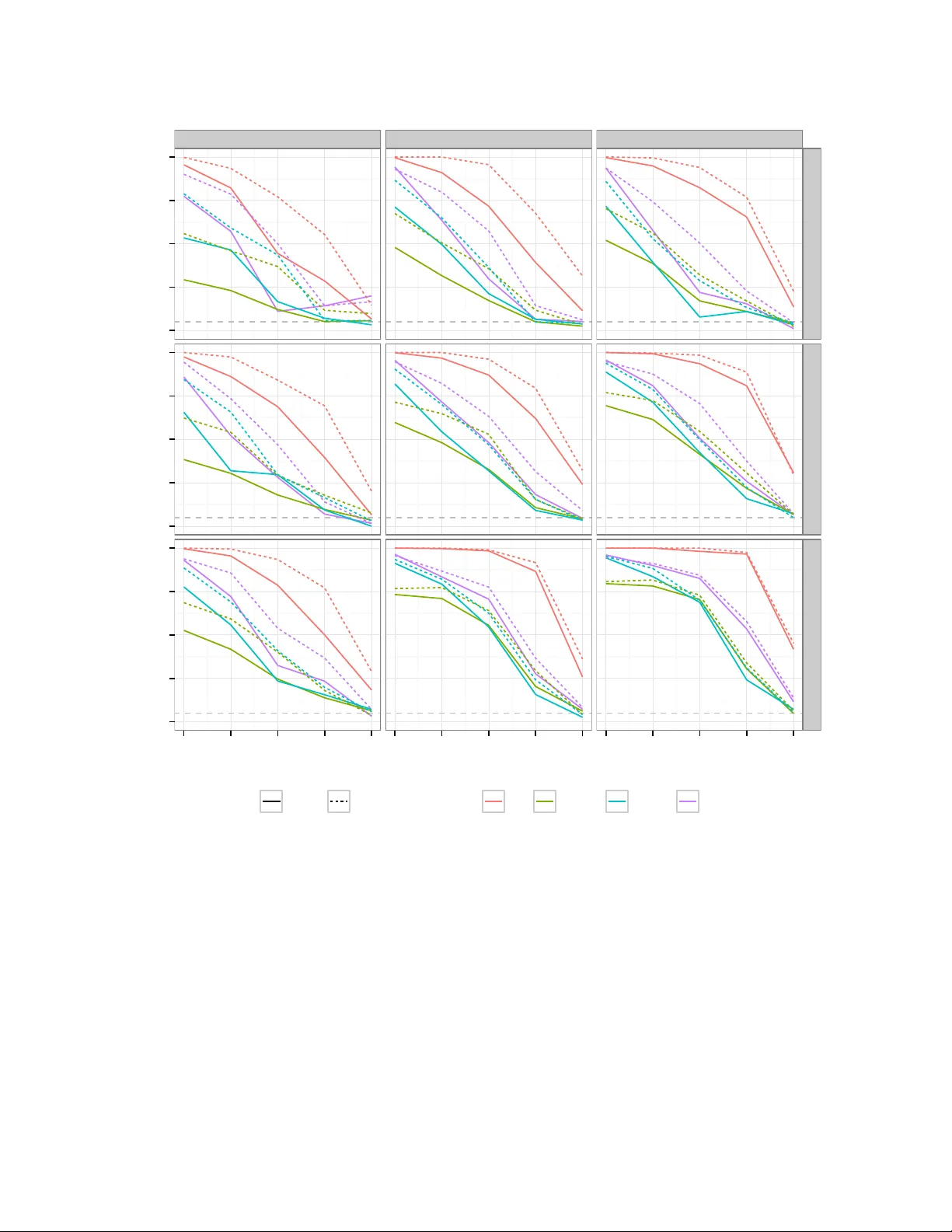
본 논문은 게놈 전역 연관 연구(GWAS)에서 서로 다른 형질이나 질환이 동일한 인과 변이에 의해 영향을 받는지를 검증하는 콜로컬라이제이션(colocalisation) 테스트의 통계적 문제점을 심도 있게 분석하고, 이를 해결하기 위한 두 가지 새로운 방법론을 제시한다.
첫 번째로, 기존 연구에서 흔히 사용되는 “가장 강하게 연관된 SNP 선택” 혹은 라쏘(LASSO)와 같은 변수 선택 절차가 실제로는 타입Ⅰ 오류를 크게 부풀린다는 점을 실증하였다. Miller(1984)의 변수 선택 편향 이론에 따르면, 변수 선택 후 동일 데이터에 회귀분석을 수행하면 회귀계수가 과대 추정되는 ‘위너스 커스’ 현상이 발생한다. 특히, LD(연관 불균형) 구조가 복잡하고 효과 크기가 작은 경우, 가장 높은 통계적 신호를 보이는 SNP이 실제 인과 SNP이 아닐 확률이 높아지며, 이는 콜로컬라이제이션 검정에서 잘못된 공유 신호를 도출하게 만든다.
두 번째로, 이러한 편향을 교정하기 위해 저자들은 두 가지 대안을 제시한다.
1) **주성분 분석(Principal Component Analysis, PCA) 기반 방법**: 전체 SNP 매트릭스를 결합해 공분산을 계산하고, 가장 큰 고유값을 가진 주성분들을 선택한다. 주성분은 서로 상관이 없으며, 선택 과정이 형질과 무관하게 이루어지므로 회귀계수에 편향이 전혀 없다. 저자는 변이의 90% 이상을 설명하는 최소한의 주성분을 사용했으며, 이는 고밀도 타깃 시퀀싱 데이터와 저밀도 GWAS 칩 모두에서 안정적인 검정력을 제공한다.
2) **베이지안 모델 평균화(Bayesian Model Averaging, BMA) 기반 방법**: 가능한 모든 SNP 조합(예: 2‑SNP, 3‑SNP 모델)에 대해 베이지안 정보 기준(BIC)으로 사후 확률을 추정하고, 각 모델별 콜로컬라이제이션 검정 p‑값을 가중 평균한다. 이 과정에서 모델 선택 불확실성을 직접 통합함으로써 변수 선택에 따른 자유도 감소와 타입Ⅰ 오류 상승을 방지한다.
시뮬레이션에서는 49개의 실제 유전적 영역을 기반으로 다양한 LD 구조와 효과 크기를 모사하였다. 결과는 기존 라쏘 기반 검정이 명목상 5% 수준의 타입Ⅰ 오류를 20~30%까지 초과시키는 반면, PCA와 BMA 방법은 모두 5% 이하로 유지하면서 검정력도 동등하거나 약간 향상됨을 보여준다. 특히, 두 방법 모두 “가장 강하게 연관된 SNP”만을 사용했을 때 발생하는 과도한 양성률을 효과적으로 억제한다.
실제 데이터 적용에서는 Graves’ disease(GD)와 Hashimoto’s thyroiditis(HT) 두 자가면역 갑상선 질환에 대한 ImmuneChip 기반 고밀도 유전체 데이터를 분석하였다. 총 13개의 위험 영역 중 7개에서는 두 질환 모두에서 동일한 인과 변이가 존재함을 확인했으며, 이는 기존 연구에서 보고된 12개 영역보다 현저히 적은 수치이다. 나머지 5개 영역은 GD에서는 강한 연관이 관찰되었지만, HT에서는 실제 연관이 없거나 검출력이 부족한 것으로 해석되었다. 이는 질환 간 공유 메커니즘을 과대평가할 위험성을 경고하고, 콜로컬라이제이션 검정의 정확한 적용이 후속 기능 연구와 치료 표적 발굴에 필수적임을 강조한다.
마지막으로, 저자들은 R 패키지 ‘coloc’를 통해 제안된 PCA와 BMA 기반 콜로컬라이제이션 검정을 손쉽게 구현할 수 있도록 제공한다. 이 패키지는 기존의 ‘coloc’ 함수와 호환되며, 사용자는 SNP 매트릭스와 형질 데이터를 입력해 자동으로 최적 주성분 수를 결정하거나, 베이지안 모델 평균화를 수행할 수 있다.
결론적으로, 본 연구는 콜로컬라이제이션 테스트의 통계적 기반을 재정립하고, 변수 선택 편향을 체계적으로 교정함으로써 유전체 연구자들이 보다 신뢰성 있게 공유 유전 신호를 탐색하도록 돕는다. 이는 향후 질환 메커니즘 해석, 기능적 후속 연구, 그리고 맞춤형 치료 전략 수립에 중요한 기여를 할 것으로 기대된다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기