Alignment-free phylogenetic reconstruction: Sample complexity via a branching process analysis
We present an efficient phylogenetic reconstruction algorithm allowing insertions and deletions which provably achieves a sequence-length requirement (or sample complexity) growing polynomially in the number of taxa. Our algorithm is distance-based, …
Authors: Constantinos Daskalakis, Sebastien Roch
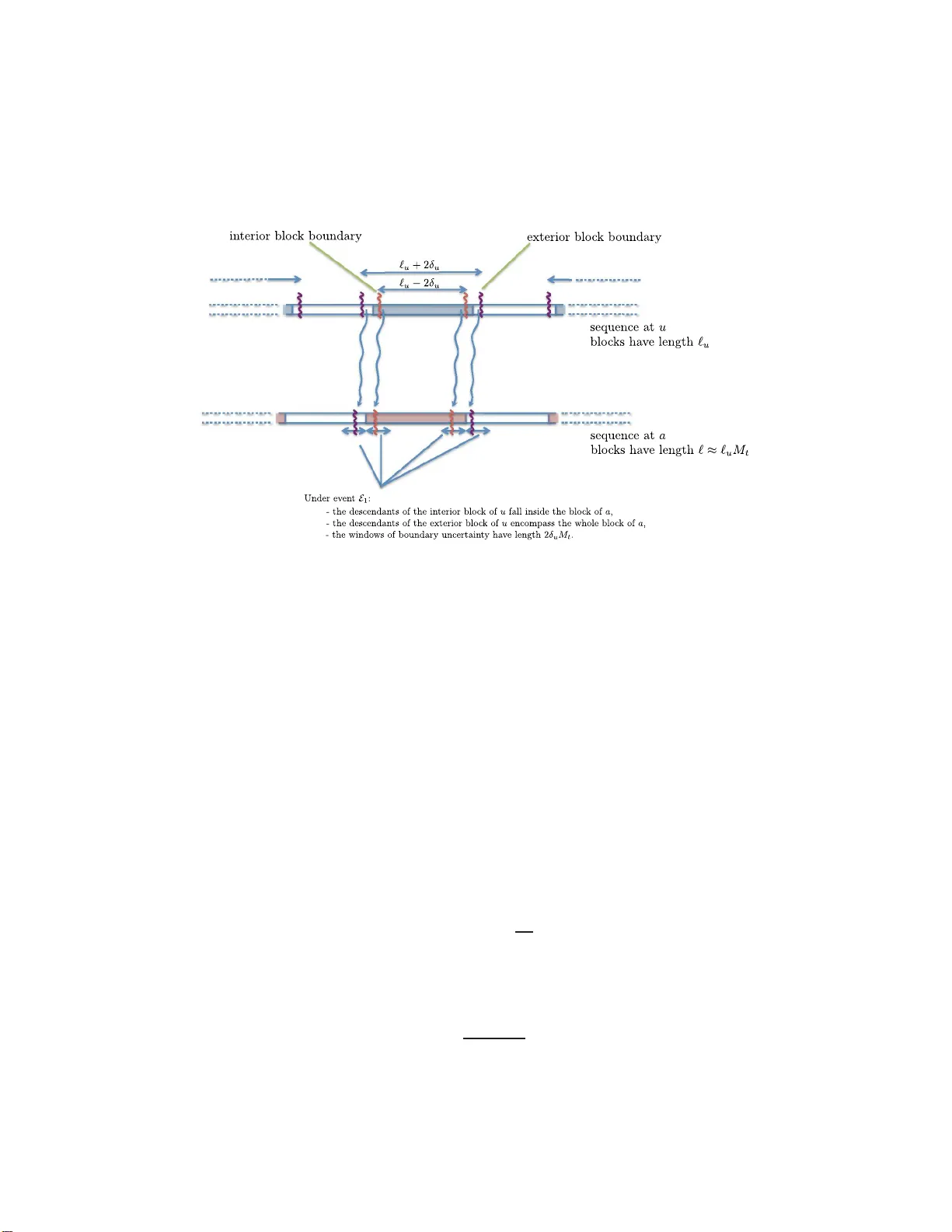
The Annals of Applie d Pr obabil ity 2013, V ol. 23, N o. 2, 693– 721 DOI: 10.1214 /12-AAP852 c Institute of Mathematical Statistics , 2013 ALIGNMENT-FREE PHYLOGENETIC RECONSTRUCTION: SAMPLE COMPLEXITY VIA A BRANCHING PROCESS ANAL YSIS By Const a n tinos D askal akis 1 and Sebas tien R och 2 Massachusetts Institute of T e chnolo gy and University of California, L os Angeles W e p resen t an efficien t phylogenetic reconstruction algorithm al- lo wing insertions and deletions which pro vably achiev es a sequence- length requirement ( or sample complexity) growing p olynomially in the num b er of taxa. Our algorithm is distance-based, t hat is, it relies on pairw ise se quence comparisons. More importantly , our approach largely bypasses the difficult p roblem of m ultiple sequence alignment. 1. In tro d uction. W e intro d uce a new efficient algorithm for the phylo- genetic tr e e r e c onstruction (PTR) problem w h ic h r igorously accoun ts for insertions and deletions. Phylo genetic b ackgr ound. A phylo genetic tr e e or phylo geny is a tree r ep- resen ting the sp eciatio n history of a group of organisms. The lea v es of the tree are t ypically existing sp ecies. The ro ot corresp onds to their most r e c ent c ommon anc estor (MR CA). Eac h branc h ing in the tree indicates a sp eciation ev ent. It is common to assume that DNA ev olv es according to a Mark ovian substitution pro cess on this phylo gen y . Under suc h a m o del, a gene is a se- quence in { A , G , C , T } k . Along eac h edge of the tree, eac h site indep enden tly m u tates according to a Marko v rate matrix. T h e length of a branc h is a measure of the amount of substitution along that branc h. Th e precise def- inition of a br anc h length dep end s on the mo del of ev olution. F or roughly constan t m utation rates, one can thin k of the branc h length as pr op ortional to the amount of time elapsed along a br anc h. The PTR pr oblem consists Received Sep tember 2011; revised F ebruary 2012. 1 Supp orted by a Sloan F oundation F ello wship and NSF Awa rd CCF-0953960 (CA- REER) and CCF-1101491. 2 Supp orted by NSF Gran t DMS-10-07144. AMS 2000 subje ct classific ations. Primary 60K35; secondary 92D15. Key wor ds and phr ases. Phylogenetic reconstruction, alignment, branching processes. This is an electronic reprint of the original article published by the Institute of Mathematical Statistics in The A nnals of Applie d Pr ob ability , 2013, V ol. 23, No. 2, 69 3–721 . This reprint differs from the orig inal in pag ination and t yp o graphic detail. 1 2 C. DASKALAKIS AN D S. ROCH of estimati ng a ph ylogen y from the g enes observ ed at its leav es. W e denote the lea v es of a tree by [ n ] = { 1 , . . . , n } and th eir sequences b y σ 1 , . . . , σ n . The mo del of sequen ce ev olution ab o v e is simplistic: it ignores man y m u- tational even ts that DNA under go es through evo lu tion. A t the gene lev el, the most imp ortant omissions are insertions and deletions of sites, also called indels . S to c hastic mo dels taking indels into accoun t ha ve long b een kno wn [ 39 , 40 ], but they are not widely used in p ractice (or in theory) b ecause of their complexit y . Instead, most practical algorithms take a t w o- phase approac h: (1) Multiple se quenc e alignment. Site t i of sequence σ i and s ite t j of se- quence σ j are said to b e homo lo gous if they descend from the same site t 0 of a common ancestor u (not necessarily th e MRCA) only thr ough substitu- tions . In the multiple se quenc e alignment (MSA) problem, we seek roughly to unco ver the homolog y relation b etw een σ 1 , . . . , σ n . T yp ically , the output is represented by a matrix D of n aligned sequences of equal length w ith v alues in { A , G , C , T , −} . Eac h column of the matrix corresp onds to h omolo- gous sites. The state − is calle d a gap and is used to accoun t for insertions and deletions. F or instance, if sequence σ l do es not hav e a site corresp ondin g to t 0 in u ab o v e, then a gap is aligned w ith p ositions t i of σ i and t j of σ j (whic h b elong to the same column). (2) Phylo ge netic tr e e r e c onstruction. T he matrix D is then cleaned up by remo ving all columns cont aining gaps. Let D ′ b e this new matrix. A stand ard PTR algorithm is then applied to D ′ . Note that s u bstitutions alone su ffi ce to explain D ′ . T raditionally , most of the researc h on phyl ogenetic metho d s has focus ed on the second ph ase. In fact, current theoretical analyses of P TR assume that the MSA problem has b een solv ed p erfe c tly . This has b een a lo ng-standing assumption in ev o- lutionary biology . But this simplifi cation is increasingly b eing questioned in the phylo genetic literature, wh ere it has b een argu ed that alignmen t heuris- tics ofte n creat e systemati c biases that affect analysis [ 26 , 42 ]. Muc h recen t empirical w ork has b een dev oted to the pr op er join t estimatio n of alignmen ts and p h ylogenies [ 25 – 28 , 32 , 37 , 39 , 40 ]. Here we giv e the fi rst analysis of an efficien t, pr o v ably consistent PTR algorithm in the presence of indels. Our new algorithm suggests th at a rough alignmen t suffices for an accurate tree reconstruction (bypassing the compu tationally difficult m u ltiple alignment problem). The or etic al pr op erties of PTR. In addition to computational efficiency , an imp ortant theoretical criterion in designing a PTR algorithm is the so- called se quenc e-length r e quir ement (SLR). At a minim um, a reconstruction ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 3 algorithm sh ould b e c onsistent , that is, assu ming a mo del of sequence ev o- lution, th e outp ut sh ould b e guarant eed to con verge on the true tree as the sequence length k (the num b er of samples ) go es to + ∞ [ 15 ]. Bey ond con- sistency , the sequence-length r equirement (or conv ergence rate) of a PTR algorithm is the sequence length required for guaran teed high-pr obabilit y re- construction. Th e SLR is t ypically giv en as an asymptotic function of n , the n u m b er of lea ves of the tree. Of co urse, it also dep end s on the s u bstitution parameters. A classical r esult due to E r d˝ os et al. [ 13 ] states that, for general trees under the assum ption that all br an ch lengths are b ounded b y constants, the so-called short quartet metho d (SQ M) has p oly ( n )-SLR. The SQM is a p articular PT R algorithm based on estimating evolutio nary distances b e- t wee n the leaf taxa, that is, th e s u m of the bran ch lengths b et we en sp ecies. Suc h algo rithms are kno wn as distanc e-b ase d metho ds . T he b asic theoreti- cal result b ehind distance-based metho ds is the f ollo wing: the collection of pairwise ev olutionary distances b etw een all sp ecies forms a sp ecial metric on th e lea ve s kno wn as an add itiv e metric; u nder mild regularit y assump - tions, su ch a metric char acterizes the und erlying phylo geny int erpreted as an edge-w eighte d tree, that is, there is a one-to-one corresp ondence b et w een additiv e metrics and p h ylogenies; moreo v er, the mapping b et wee n them can b e computed efficiently [ 3 4 ]. A new app r o ach. In the classical theoretical setting ab o ve where the MSA problem is assumed p erfectly solv ed (we refer to this setting b elo w as the ESSW framew ork), the ev olutionary d istance b et w een t w o sp ecie s is measured using the Hamming d istance (or a s tate-dep endent generalizatio n) b et w een their resp ectiv e sequences. It can b e s h o wn that after a prop er correction f or multiple sub stitutions (which d ep ends on th e mo del used) the exp ectation of the quan tit y obtained do es satisfy the additiv e metric prop erty and can therefore serv e as the basis for a distance-based PTR algorithm. Mo ving b eyo n d the ESSW fr amework, it is tempting to account for indels b y simply usin g edit distance instead of the Hamming d istance. Reca ll that the e dit distanc e or L evenshtein distanc e b etw een t wo str in gs is give n by the minimum n u m b er of op erations n eeded to tr an s form one strin g in to the other, wh ere an op eration is an insertion, deletion or sub stitution of a s ingle c haracter. Ho wev er, no analytical exp ression is kno wn for the exp ecta tion of edit distance under standard indel mo d els and compu ting such an expr ession app ears difficult (if at all p ossible). An alternativ e idea is to compute the maximum likeliho o d estimator for the time elapsed b et w een tw o sp ecies give n their sequences. But this inv olv es solving a nonconv ex optimization problem and the lik eliho o d is only kn o wn to b e efficien tly computable under a r ather unrealistic assumption k n o wn as rev ersibility [ 39 ] (see b elo w). 4 C. DASKALAKIS AN D S. ROCH W e use a different approac h. W e d ivide the s equ ences int o quantile blo c ks (the fir st x % , the second x % , etc.). W e sho w that by appropriately choosing x ab o v e we can mak e su re that the b lo c ks in different sequences essen tially “matc h” eac h other, that is, they are m ade of mostly homologo us sites. W e then compare the s tate frequencies in matc h ing blocks and build an additiv e metric out of this s tatistic. As we sh o w b elo w, this is in fact a natural generalization of the Hamming estimator of the ESSW framework. Ho we v er, unlike the Hamming distance w hic h can easily b e analyzed through standard concen tration inequalities, pr o ving rigorously that our app roac h w orks in volv es sev eral new tec hn ical difficulties. Our analysis relies on a branc hing pro cess analysis of the site displacemen ts. W e giv e a quic k pro of sk etc h after the formal statemen t of our results in Section 1.2 . The resu lts d escrib ed here were fi r st announ ced without pro of in th e sp e- cial case of u ltrametric trees u nder the CFN mo del with inv er s e logarithmic indel rates [ 10 ]. Here w e g iv e full pro ofs of stronger results, includin g exten- sions to b ounded-rate trees und er GTR mo dels. R elate d work. F or more b ac kground on mo dels of m olecular ev olution and phyloge netics, see, for example, [ 16 , 17 , 34 ]. F ollo wing the s emin al results of [ 13 ], there has b een muc h work on sequence-length requirement, including [ 4 – 9 , 14 , 18 , 20 , 23 , 24 , 29 , 30 , 33 , 35 , 36 ]. The multiple sequence alignmen t problem as a com binatorial optimizat ion problem (fin ding the b est alignmen t un d er a giv en pairwise s coring fu nction) is kno wn to b e NP-hard [ 12 , 41 ]. Most heur istics used in practice, su ch as CLUST AL [ 19 ], MAFFT [ 22 ] and MUSC LE [ 11 ], u se the id ea of a guide tree, that is, they firs t construct a v ery rough phyloge netic tree from th e data (using, e.g., edit d istance as a measure of ev olutionary distance), and then recursiv ely co nstruct lo cal alignmen ts pro du ced by “aligning alignmen ts.” T o our kno wledge, little theoretical w ork has b een dedicated to the join t estimation of alignmen ts and p h y logenies, w ith the exception of Th atte [ 38 ] who ga ve consistency resu lts for the rev er s ible case in the limit where the deletion-to-i nsertion ratio tends to 1. Ho w ev er, no sequence-length require- men t is obtained in [ 38 ]. In recen t r elated w ork, the pr oblem of reconstruct- ing ancestral sequences in the presence of indels w as considered [ 1 , 2 ]. 1.1. Mo del of se quenc e e volution. Phylo geny. A phylo geny is represented b y a bin ary tree T = ( V , E ), whose lea v es L ⊂ V corresp ond to extan t sp ecies, and whose bifur cations denote ev olutionary even ts whereby t wo new sp ecies are generated f rom an ancestor. The ro ot of the ph y logeny , denoted by r ( T ), r epresen ts the common ancestor of all the sp ecies in the phylo geny , and we assume that all edges of T are directed a w ay from r ( T ) ; so, if e = ( u, v ) is a b ranc h of the phylogen y , u is the p ar ent of v and v is the child of u . Moreo ver, if v ′ is in the subtree of T ro oted at u , w e call v ′ a desc endant of u and u an anc e stor of v ′ . ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 5 Along eac h b ranc h of the phylogen y , the genetic material of the paren t sp ecies is sub j ect to mo d ifications that pro duce the genetic material of its c hild sp ecies. A common biological assumption is that th e genetic m aterial of eac h sp ecies u can b e represente d b y a binary sequence σ u = ( σ 1 u , . . . , σ K u u ) of length K u o ve r a fi n ite alphab et—for ease of present ation, we w ork with a bi- nary alphab et { 0 , 1 } (bu t see Section 5 for extensions to ric her alphab ets)— and that the c hanges to whic h σ u is sub jected along the br anc h e = ( u, v ) are describ ed b y a Marko v pro cess. In particular, the Mark o v pr op ert y im- plies th at, giv en the s equ ence σ u of u , the sequ ence σ v is in dep end en t of the sequences of the sp ecie s outside the subtree of T ro oted at u . A simp lifying assu m ption commonly used in phylog enetics is th at all sp ecies hav e sequences of the same length and , moreo ve r , that ev ery site , that is, ev ery co ord inate, in their s equences evo lv es in dep end en tly f rom ev ery other site. In particular, it is assu med that, along eac h branch e = ( u, v ) of the phylog en y , ev ery s ite σ j u of the sequen ce σ u is flipp ed with probabilit y p e to the v alue 1 − σ j u indep en d en tly from the other sites. This mo del is kn o wn as the Ca v ender–F arris–Neyman (CFN) m o del. A simp le generalizatio n to { A , G , C , T } is known as the Juk es–Cant or (JC) model (see, e.g. , [ 16 ]). A c c outing for indels. In this pap er, we consider a more general evo lu- tionary pro cess that acc oun ts for the p ossibilit y of insertions and d eletions. Our mo del is similar to the original TKF91 mo del [ 39 ], except that we do not enforce rev ersibilit y . In our mo d el, ev ery edge e = ( u, v ) of the phylogen y is c haracterized b y a quadruple of parameters ( t e ; η e , µ e , λ e ), wh ere t e is the ev olutionary time b et w een the sp ecies u and v , and η e , µ e and λ e are, resp ec- tiv ely , the substitution , deletion and insertion rates. The Marko v process b y whic h the sequence at v is obtained from the sequence at u is defi ned b elo w (see, e.g., [ 21 ] for b ackg round on c on tinuous-time Mark o v p r o cesses). Definition 1.1 (Ev olutionary pr o cess on a branc h ). G iv en an edge e = ( u, v ) , with paramete r s ( t e ; η e , µ e , λ e ), the sequ ence σ v at v is obtained f rom the sequence σ u at u acc ording to th e follo win g Mark o v pro cess: (1) Intia lize σ v := σ u , K v := K u and t ℓ := t e (where t ℓ is the r emaining time on the edge e ). (2) While t ℓ > 0: • ( Timing of next e vent ) let I 0 , I 1 , . . . , I K v b e exp onen tial random v ari- ables with rate λ e , D 1 , . . . , D K v exp onentia l r an d om v ariables with rate µ e and M 1 , . . . , M K v exp onentia l r andom v ariables with rate η e ; supp ose that these rand om v ariables are m utually in dep end en t and let T b e their minimum; • if T > t ℓ , th e pro cess ends at t ℓ ; otherwise: – ( Insertion ) if I j = T , insert a new site wh ose v alue is c hosen u ni- formly at random from { 0 , 1 } b etw een the sites σ j v and σ j +1 v of σ v ; 6 C. DASKALAKIS AN D S. ROCH – ( Deletion ) if D j = T , delete the site σ j v from σ v ; – ( Substitution ) and if M j = T , replace σ j v b y 1 − σ j v ; (If j = 0, then σ j v is und efined and, if j = K v , then σ j +1 v is u ndefined .) • ( R emaining time ) u p d ate σ v according to th ese changes, and up date K v to r eflect the new sequence length; set the remaining time t ℓ := t ℓ − T . In words, th e ev olutionary pro cess defined ab ov e assumes that ev ery site of the sequence σ u of the parent sp ecies is, indep end en tly from the other sites, sub ject ed to a sequence of ev olutionary eve nts that fl ip its v alue; these ev ents are distributed according to a P oisson p oin t p ro cess of int ensit y η e in the time int erv al [0 , t e ]. Ho wev er, the site may get d eleted and th erefore not b e inherited by the sequence of the no de v ; this is determined by whether an exp onen tial random v ariable of rate µ e is sm aller than t e . While eac h site of the paren tal sequence σ u is sub jected to th is pro cess, n ew sites are in tro duced in th e space b et we en existing sites at rate λ e , and eac h of these sites follo w s a similar pro cess for the remaining time. In essence, insertion and deletion eve n ts are gov erned b y an in dep end en t br anc hin g p ro cess for eac h ancestral site. Note f u rther that the order of the sites, as describ ed ab o ve, also plays a role. Remark 1.2. Unlik e [ 39 ], we do not use an “ immortal link” and we do not assum e that the length pro cess is at stationarit y . Ou r tec hn iqu es can also b e ap p lied to th e TKF 91 mod el with ou t m uch mo difications. W e lea v e the details to the reader. Giv en the evo lu tionary pro cess on a br anc h of the ph ylogen y , the ev olu- tionary pro cess o n the w hole ph ylogen y is defined as follo w s . Definition 1.3 (Ev olutionary pro cess). Supp ose that ev ery site of the sequence σ r ( T ) at the ro ot of the phylogen y is c hosen to b e 0 or 1 uniformly at random. Recursiv ely , if σ u is th e sequen ce at no de u and e = ( u, v ) is an edge of the ph ylogen y , the sequence σ v at no de v is obtained from the sequence σ u b y an application of the ev olutionary pro cess on a branch describ ed by Definition 1.1 . F or ease of exp osition, w e first p resen t our pro of in the sp ecial case w here the substitution, insertion and deletion rates are the same on all edges of the ph ylogeny . Definition 1.4 (Ultrametric assu mption). Under the ultrametric as- sumption, the lea v es of the phylo gen y are con temp oraneous, that is, there exists H su c h that f or eac h u ∈ L the su m of ev olutionary times t e on the branc hes betw een u and the ro ot is H . ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 7 Definition 1.5 (Molec ular clock assumption). Under the molec u lar clock assumption, we assume that the ultrametric assumption h olds . More- o ve r, there exist η , µ and λ suc h that η e = η , µ e = µ and λ e = λ , for all e ∈ E . W e discuss a m ore general case in Sect ion 5 . Notation. In the sequel, w e lab el the lea ves of the ph y logeny w ith the p ositiv e in tegers 1, 2 , . . . , n , so that L = { 1 , . . . , n } , a nd the root r ( T ) of the phylo geny with 0. 1.2. Main r esult. Statement of r esu lts. W e b egin with a consistency r esult. Here we con- sider a completely general p h ylogen y , that is, neither the ultrametric n or the molecular clock assumptions need hold. Theorem 1 (Consistency: finite case). Assume that 0 < t e , η e , λ e , µ e < + ∞ , for al l e ∈ E . Then ther e exists a pr o c e dur e r etu rning the c orr e ct tr e e fr om the se quenc es at the le aves, with pr ob ability of failur e appr o aching 0 as the se quenc e leng th at the r o ot of the tr e e go es to + ∞ . Our main r esult is the follo wing. F or simplicit y we fi rst work u nder the symmetric tw o-state case and assu me that the molecular clo c k assumption holds. Theorem 2 (Main r esult: tw o-state, molecular clo c k case). Consider the two-state mo del under the mole cular clo ck assumption. Assume further that ther e exist c onstants 0 < f , g < + ∞ , indep endent of n , su ch that f < t e < g ∀ e ∈ E . Mor e over, assume that η e = η, λ e = λ, µ e = µ ∀ e ∈ E , wher e η , λ and µ ar e b ounde d b e twe en c onstants (indep endent of n ) 0 < ¯ η < ¯ η < + ∞ , 0 = ¯ λ < ¯ λ < + ∞ and 0 = ¯ µ < ¯ µ < + ∞ , r esp e ctively. Under the assumptions ab ove, for al l β ′ > 0 ther e exists β ′′ > 0 such that ther e e xists a p olynomial-time algorith m solving the phylo ge netic r e c onstruction pr oblem (i.e., r eturning the c orr e ct tr e e) with pr ob ability of failur e n − β ′ , i f the r o ot se quenc e has length k r ≥ n β ′′ . 3 3 In [ 10 ], a preliminary vers ion of this result was announced without pro of, with the muc h stronger assumption that ¯ λ, ¯ µ = O (1 / log n ) , that is, th at the indel rates are negligi- ble. Here we sho w that th is assumption can b e relaxed (at the cost of longer sequences). 8 C. DASKALAKIS AN D S. ROCH Remark 1.6 (Branch lengths). Our assumption that all b ranc h lengths t e , e ∈ E , satisfy f < t e < g is standard in the sequence-length r equirement literature follo wing the seminal work of [ 13 ]. Extensions. In S ection 5 w e d eriv e the follo w ing extension. Let Q b e a rev ersib le 4 × 4 rate matrix with stationary distrib u tion π . (Larger alphab ets are also p ossible.) T he GTR sequence evol ution pro cess is identica l to the one d escrib ed in Definition 1.1 except that the substitution pr o cess is a con tinuous-time Ma rk ov pro cess w ith rate mat rix η e Q . Theorem 3 (Main result: GTR, b oun ded-rates case). Consider the GTR mo del with r ate matrix Q under the ultr ametric assumption (but not ne c- essarily the mole cular clo ck assumption) . Assume further t hat th er e exist c onstants 0 < f , g , ¯ η , ¯ η , ¯ λ, ¯ λ, ¯ µ, ¯ µ, < + ∞ , indep endent of n , su ch that f < t e < g , ¯ η < η e < ¯ η ∀ e ∈ E . Mor e over, assume that λ e = λ, µ e = µ ∀ e ∈ E , wher e λ and µ ar e b ounde d b etwe en c onstants (indep endent of n ) 0 = ¯ λ < ¯ λ < + ∞ and 0 = ¯ µ < ¯ µ < + ∞ , r esp e ctively. We r ef er to the c onditions ab ove as the b ounde d-r ates assumption. Under the assumptions ab ove, for al l β ′ > 0 ther e exists β ′′ > 0 such that ther e exists a p olynomial-time algorithm solving the phylo genetic r e c onstruction pr oblem (i.e., r eturning the c orr e ct tr e e ) with pr ob ability of failur e n − β ′ , if the r o ot se quenc e has length k r ≥ n β ′′ . Pr o of sketch. Consider the t w o-state, molecular clo c k ca s e. As we noted b efore, un like the classical setting wh ere the Hamming distance ca n b e an- alyzed through standard concen tration inequalities, p ro vin g rigorously th at our app r oac h works in v olve s sev eral new tec hnical difficulties. The pro of go es through the follo wing steps: (1) Exp e ctations. W e fi rst compute exp ectations of blo c k statistics, which in volv e analyzing a cont in u ous-time Mark o v pro cess. W e use these calcula- tions to d efine an appropriate add itiv e metric based on correlations b et ween blo c ks . (2) Se quenc e length and site displac ements. W e giv e b oun ds on ho w muc h sequence lengths v ary across the tree through a moment-g enerating fun ction argumen t. Usin g our b ounds on the sequence length p ro cess, w e b ound the w orst-case displacemen ts of the sites. Namely , w e sh o w th at, under our as- sumptions, all sites mo ve b y at most O ( √ k log k ). ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 9 (3) Se quenc e p artitioning. W e divide eac h sequ ence in b lo c ks of size r ough- ly k ζ for ζ > 1 / 2, where k is the sequence length at th e ro ot. F rom our b ound s on site d isplacemen ts, it follo ws that th e blo c ks rough ly m atch across differen t sequences. In p articular, we b ound th e num b er of homologous sites b et w een matc hing blocks with high probabilit y and sh o w that the exp ected correlation b et wee n these blocks is appro ximately correct. (4) Conc entr ation. Finally , we sh ow th at our estimates are concentrat ed. The concen tration argument pro ceeds by co nditioning on th e indel pro cess satisfying the h igh-probabilit y conditions in the pr evious p oin ts. The crux of our result is the p r op er estima tion of an additive m etric. With suc h an estimatio n pro cedure in hand, w e can use a standard distance-based approac h to reco v er the ph ylogen y . Or ganization. The rest of the pap er is organized as follo ws. The evo lu- tionary d istance forming the basis of our approac h is pr esen ted in Section 2 . W e describ e our fu ll distance estimator in Section 3 and p ro ve its concen- tration in the same sectio n. Extensions are d escrib ed in Section 5 . 2. Ev olutionary distances. Consider the t wo -state, m olecular clo c k case. In this section, we s ho w ho w to define an appropr iate notion of “ev olutionary distance” b et w een t wo sp ecies. Although such distances ha ve b een widely used in prior p h ylogenetic w ork and h a ve b een defined for a v ariet y of mo dels [ 16 , 34 ], to our knowle dge our definition is the first that applies to mo dels with indels. W e b egin by reviewing the standard definition in the indel-free case and then adap t it to the presence of indels. Our estimation pr o cedure is discussed in Section 3 . 2.1. The classic al indel-f r e e c ase. Supp ose first that λ = µ = 0, that is, ther e is no indel . In that case, the sequence length remains fixed at k and the alignmen t problem is trivial. Underlyin g all d istance-based app roac hes is the follo w ing basic definition. Definition 2.1 (Additiv e metric). A phylog en y is naturally equipp ed with a so-called additiv e metric on the lea ves D : L × L → (0 , + ∞ ) defined as ∀ a, b ∈ L D ( a, b ) = X e ∈ P T ( a,b ) ω e , where P T ( a, b ) is the set of edges on th e path b et w een a and b in T and where ω e is a nonnegativ e function of the p arameters on e (in our case, t e , η e , λ e and µ e ). F or instance, a common c hoice for ω e w ould b e ω e = η e t e in whic h case D ( a, b ) is the exp ected n umb er of su bstitutions p er site b et w een a a nd b . Often D ( a, b ) is referred to as the “ev olutionary distance” b et w een sp ecies a and b . Add itiv e metrics are c h aracterized by the follo win g four - 10 C. DASKALAKIS AN D S. ROCH p oint condition: for al l a, b, c, d ∈ L , D ( a, b ) + D ( c, d ) ≤ max {D ( a, c ) + D ( b, d ) , D ( a, d ) + D ( b, c ) } . Moreo v er, assumin g ω e > 0 for all e ∈ E , it is w ell kno wn that th er e exists a one-to-one corresp ondence b et we en D and T as a weigthed tree with edge w eights { ω e } e ∈ E . W e will discuss algorithms for constructing T from D in Section 4 . F or more bac k grou n d on tree- based metrics, see [ 34 ]. Definition 2.1 implies that ph ylogenies can b e reconstru cted b y comput- ing D ( a, b ) for all pairs of lea ves a, b ∈ L . Assume we seek to estimate the ev olutionary distance b etw een sp ecies a and b using their resp ectiv e se- quences. In a first attempt, one might try the (norm alized) Hamming dis- tance b et we en σ a = ( σ 1 a , . . . , σ k a ) and σ b = ( σ 1 b , . . . , σ k b ). How ev er, the exp ected Hamming distance (in other words, th e probability of disagreemen t b et w een a site of a and b ) do es not form an additive m etric as defin ed in Defini- tion 2.1 . Instead, it is w ell kn own that an appropr iate estimator is obtained b y “correcting” the Hamming distance for “m ultiple” substitutions. De not- ing by b H ( σ a , σ b ) the Hammin g distance b etw een σ a and σ b , a Mark ov c h ain calculatio n sho w s that D ( a, b ) = − 1 2 log(1 − 2 E [ b H ( σ a , σ b )]), with th e choice ω e = η e t e (see, e .g., [ 16 ]). In a distance- based reco nstruction pro cedur e, one first estimate s D with b D ( a, b ) = − 1 2 log(1 − 2 b H ( σ a , σ b )) (1) and then app lies one of the algorithms discussed in S ection 4 b elo w. T h e sequence-length requirement of suc h a metho d can b e d er ived by using con- cen tration results for b H [ 4 , 13 ]. 2.2. T aking indels into ac c ount. T o simplify the pr esentati on, w e assume throughout that λ 6 = µ . The case λ = µ follo ws from the same argumen t. In the presence of indels, the estimator ( 1 ) based on the Hamming distance is d ifficult to app ly . One has to first align th e sequen ces, whic h cannot b e done p erfectly and causes b iases as well as correlations that are hard to analyze. Alte rnativ ely , one could try a differen t string distance suc h as edit distance. Ho wev er, computing the exp ectat ion of edit distance und er indel mo dels app ears difficult. W e use a differen t approac h inv olving correlations b etw een state frequen- cies. W e will ev en tually app ly th e estimator to large sub-blo cks of the se- quences (see Section 3 ), b ut w e first describ e it for the full sequence for clar- it y . F or a no de u , let K u b e the (random) length of the sequence at u and Z u , th e num b er of 0’s in the sequence at u . Then, our distance estimator is b D ( a, b ) = ( Z a − 1 2 K a )( Z b − 1 2 K b ) . W e now analyze the expectation of this quantit y . F or u ∈ V , w e let ∆ u = Z u − 1 2 K u ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 11 b e the d eviation of Z u from its exp ected v alue (conditioned on the sequence length). Single c hannel. Supp ose T is made of a single edge fr om the ro ot r to a leaf a with parameters t, η , λ, µ . Assume first that the original sequence length is k r = 1 . Let K a b e the length of the sequence at a . Th en K a is a con tinuous-time bran ching pro cess and, by Marko v chain calculations ([ 3 ], Section I I I.5), its moment- generating function is F ( s, t ) ≡ E [ s K a ] = µ ( s − 1) − e ( µ − λ ) t ( λs − µ ) λ ( s − 1) − e ( µ − λ ) t ( λs − µ ) . (2) By differen tiating F ( s, t ) we derive E [ K a ] = e − ( µ − λ ) t (3) and V ar[ K a ] = µ + λ µ − λ [ e − ( µ − λ ) t − e − 2( µ − λ ) t ] . (4) Let K ∗ a b e the n u mb er of “new” sites at a , that is, excluding the original site if it surv ived. (W e ignore the su bstitutions for the time b eing.) The probabilit y that the original site survive s is e − µt . Then, E [ K ∗ a ] = E [ K a − 1 { original site survive s } ] = e − ( µ − λ ) t − e − µt b y linearit y of expectation. W e no w tak e into accoun t sub s titutions. Assume th at the original se- quence length at r is a ran d om v ariable K r and that the sequence at r is i.i.d. uniform. Denote b y Z r the n um b er of 0’s at r . T he probabilit y that a site in r , that is still sur v ivin g in a , has flipp ed its v alue is p = P [state flips odd n umb er of times in time t ] = + ∞ X j =0 e − ηt ( η t ) 2 j + 1 (2 j + 1)! = e − ηt sinh η t = 1 − e − 2 ηt 2 . Also, n ote that a new site created along the path b et w een r and a has equal c hance of b eing 0 or 1 at the end of the p ath . Then we ha v e the follo w ing lemma. Lemma 2.2 (Sin gle c h annel: exp ected deviatio n). The fol lowing holds: E [∆ a | K r , Z r ] = e − (2 η + µ ) t ∆ r . 12 C. DASKALAKIS AN D S. ROCH Pr oof. W e ha ve E [∆ a | K r , Z r ] = E [( Z a − 1 2 K a ) | K r , Z r ] = Z r e − µt (1 − p ) + ( K r − Z r ) e − µt p + K r ( e − ( µ − λ ) t − e − µt ) 1 2 − K r e − ( µ − λ ) t 1 2 (5) = Z r (1 − 2 p ) e − µt − 1 2 K r (1 − 2 p ) e − µt = e − 2 ηt e − µt ∆ r , where on the first t wo lines: (1) the first term is the num b er of original 0’s surviving in state 0; (2) the seco nd term is the n u m b er of original 1’s survivin g in state 0; (3) the thir d term is the num b er of new sites surviving in state 0 (where recall that n ew site s are uniformly c h osen in { 0 , 1 } ); (4) the fourth term is half the sequence length at a giv en the length at r . F ork channel. Consider n o w a “fork” tree, that is, a ro ot r f rom which emanates a single edge e u = ( r, u ) wh ic h in turn branches into t wo edges e a = ( u, a ) and e b = ( u, b ) (see Figure 1 b elo w). F or x = a, b, u , w e denote the parameters of edge e x b y t x , λ x , µ x , η x . Our goal is to compute E [ b D ( a, b )] assuming that the s equence length at the ro ot is k r . W e u se ( 5 ), th e Mark ov prop erty and the fact th at Z u conditioned on K u is a binomial with param- eters 1 / 2 and K u . W e get th e f ollo wing lemma. Lemma 2.3 (F ork c hannel: exp ected distance). The fol lowing holds: E [ b D ( a, b )] = e − (2 η a + µ a ) t a e − (2 η b + µ b ) t b e − ( µ u − λ u ) t u k r 4 . Pr oof. W e ha ve E [ b D ( a, b )] = E [∆ a ∆ b ] = E [ E [∆ a ∆ b | K u , Z u ]] = E [ E [∆ a | K u , Z u ] E [∆ b | K u , Z u ]] = e − 2 η a t a e − µ a t a e − 2 η b t b e − µ b t b E [∆ 2 u ] = e − 2 η a t a e − µ a t a e − 2 η b t b e − µ b t b E [ E [∆ 2 u | K u ]] = e − 2 η a t a e − µ a t a e − 2 η b t b e − µ b t b E K u 4 = e − 2 η a t a e − µ a t a e − 2 η b t b e − µ b t b e − ( µ u − λ u ) t u k r 4 , where w e used ( 3 ) and Lemma 2.2 . ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 13 Mole cular clo ck . W e s p ecialize the previous result to the molecular clo c k assumption. T h at is, w e assume, for x = a, b, u , that λ x = λ , µ x = µ and η x = η . Note that by construction t a = t b (assuming sp ecies a and b are con temp orary). W e denote t = t a and ¯ t = t u + t a . Denoting κ = k r e − ( µ − λ ) ¯ t 4 , w e then get the follo wing lemma. Lemma 2.4 (Molecular clo c k: exp ected d istance). The fol lowing holds : E [ b D ( a, b )] = e − (4 η + µ + λ ) t κ. Letting β = 4 η + µ + λ, w e get that − 2 log E [ κ − 1 b D ( a, b )] = 2 β t, whic h is the ev olutionary d istance b etw een a and b with the c hoice ω e = β t e . Therefore, w e define the follo wing estimator: b D ∗ ( a, b ) = − 2 log κ − 1 b D ( a, b ) . 3. Distance computation. W e no w show h o w to estimate the ev olution- ary distance b et w een t wo sp ecies b y decomp osing the sequ ences in to large blo c ks which serv e as r oughly indep en d en t samples. W e use the follo wing no- tation: M t = e − ( µ − λ ) t , D t = e − µt , δ = µ − λ , φ = µ + λ and Γ t = δ − 1 λ (1 − M t ). W e sh o w in Section 4 that th e time elapsed b etw een the ro ot and the lea v es is b ounded b y g 2 f log 2 n . Hence, under our assumptions Υ − 1 n ≡ e − ( ¯ µ + ¯ λ )( g 2 /f ) log 2 n ≤ e − ( ¯ µg 2 /f ) log 2 n (6) ≤ M t ≤ e ( ¯ λg 2 /f ) log 2 n ≤ e ( ¯ λ + ¯ µ )( g 2 /f ) log 2 n ≡ Υ n , Υ − 1 n ≤ e − ( ¯ µg 2 /f ) log 2 n ≤ D t ≤ 1 (7) and 0 ≤ Γ t = λt 1 − e − ( µ − λ ) t ( µ − λ ) t ≤ ¯ λ g 2 f log 2 n e ( ¯ λg 2 /f ) log 2 n − 1 ( ¯ λg 2 /f ) log 2 n (8) = e ( ¯ λg 2 /f ) log 2 n − 1 ≤ Υ n , where we used that the function x − 1 (1 − e − x ) is nonnegativ e and decreasing since its d eriv ativ e is xe − x − (1 − e − x ) x 2 = e − x (1 + x ) − e x x 2 ≤ 0 , x 6 = 0 . Note that th e b ou n ds ab o ve are p olynomials in n with exp onen ts dep ending only on f , g , ¯ λ and ¯ µ . In particular, w e will ultimately take s equ ence lengths 14 C. DASKALAKIS AN D S. ROCH k r of the form n β ′′ with β ′′ c hosen muc h larger than the exp onen t in Υ n . W e call p olynomials in n (suc h as Υ n ) which ha ve an exp onent not dep ending on β ′′ , smal l p olynomials . As a result, the follo wing notation will b e useful. F or a f unction W ( k r ) of k r , we use S n ( W ( k r )) to denote a fun ction smaller or e qual to W ( k r ) up to a smal l p olynomial factor . (The la tter will b e used similarly to the big-O notati on.) Recall the f ollo wing standard conce n tr ation inequ alities (see, e.g., [ 31 ]). Lemma 3.1 (Cher n off b ounds). L et Z 1 , . . . , Z m b e indep endent { 0 , 1 } - r andom variables such that, for 1 ≤ i ≤ m , P [ Z i = 1] = p i wher e 0 < p i < 1 . Then, for Z = P m i =1 Z i , M = E [ Z ] = P m i =1 p i , 0 < δ − ≤ 1 and 0 < δ + ≤ U , P [ Z < (1 − δ − ) M ] < e − M δ 2 − / 2 and P [ Z > (1 + δ + ) M ] < e − c ( U ) M δ 2 − , wher e c ( U ) = [(1 + U ) ln(1 + U ) − U ] /U 2 . 3.1. Conc e ntr ation of the indel pr o c ess. Se quenc e length. W e fir st sho w that the sequen ce length is concentrat ed. Let T b e single c hannel consisting of edge e = ( r , a ). Let k r b e the length at r . Lemma 3.2 (Sin gle c hann el: large deviations of sequence length). F or al l γ > 0 and b k r ≥ k r = n β ′′′ with β ′′′ > 0 lar ge enough, with pr ob ability at le ast 1 − b k − γ r , K a = k r M t ± S n ( q b k r log b k r ) , wher e the smal l p olynomial factor in S n ( q b k r log b k r ) dep ends on γ as wel l. Remark 3.3. Although we stated Lemma 3.2 for the full sequen ce, it will also b e needed for “half-sequences” and “blo c ks.” In particular, w e u se the p revious lemma to trac k the p osition of sites. In that con text, one should think of k r as th e p osition of a site in r and K a as its p osition in a . Then w e can use b k r for the fu ll s equence length at r (see Sectio n 3.2 ). Pr oof of Lemma 3.2 . W e think of K a as K a = k r X i =1 K a,i , where K a,i is the num b er of sites generated by a sin gle site of the sequence at r . Intuitiv ely , K a,i is the n umber of sites that w ere inserted b et ween the ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 15 sites i and i + 1 of the sequence at r , plu s the site at p osition i itself, if it survive d. Clea rly the v ariables { K a,i } i are m utually indep en d en t. Using ( 3 ) we obtain that E [ K a ] = k r M t . F or ε > 0, b y Mark ov’s inequalit y , w e ha v e P [ K a ≥ k r M t + k r ε ] ≤ s − k r ( M t + ε ) E [ s K a ] = ( s − ( M t + ε ) E [ s K a, 1 ]) k r . (9) W e tak e s = 1 + C ε for C > 0 to b e determined. W e hav e E [ s K a, 1 ] = µ ( s − 1) − e ( µ − λ ) t ( λs − µ ) λ ( s − 1) − e ( µ − λ ) t ( λs − µ ) = ( µ − λM − 1 t ) C ε + δ M − 1 t λ (1 − M − 1 t ) C ε + δ M − 1 t = δ − 1 ( µM t − λ ) C ε + 1 δ − 1 λ ( M t − 1) C ε + 1 = 1 − ( λ − 1 µ Γ t − 1) C ε 1 − Γ t C ε = [1 − ( λ − 1 µ Γ t − 1) C ε ] + ∞ X ι =0 [Γ t C ε ] ι , whenev er Γ t C ε < 1 . Hence, if Υ n C ε < 1 is b ounded a w a y fr om 1 (indep en- den tly of n ), w e ha v e, using ( 8 ), E [ s K a, 1 ] = [1 − ( λ − 1 µ Γ t − 1) C ε ][1 + Γ t C ε + (Γ t C ε ) 2 + O ((Υ n C ε ) 3 )] = 1 + M t ( C ε ) + M t Γ t ( C ε ) 2 + O ((Υ n C ε ) 3 ) . Moreo v er, using the binomial series and ( 6 ), and assuming C ε < 1 s − ( M t + ε ) = + ∞ X ι =0 ( − M t − ε )( − M t − ε − 1) · · · ( − M t − ε − ι + 1) ι ! [ C ε ] ι ≤ 1 − ( M t + ε )( C ε ) + ( M t + ε )( M t + ε + 1) 2 ( C ε ) 2 + + ∞ X ι =3 ( M t + ε + 1) ι [ C ε ] ι = 1 − ( M t + ε )( C ε ) + ( M t + ε )( M t + ε + 1) 2 ( C ε ) 2 + O ((Υ n C ε ) 3 ) , whenev er ε is small and Υ n C ε < 1 is b oun ded a wa y from 1 (ind ep endently from n ). Therefore, s − ( M t + ε ) E [ s K a, 1 ] = 1 − ε ( C ε ) + M t Γ t ( C ε ) 2 + ( M t + ε )( M t + ε + 1) 2 ( C ε ) 2 − ( M t + ε ) M t ( C ε ) 2 + O ((Υ n C ε ) 3 ) . 16 C. DASKALAKIS AN D S. ROCH Fig. 1. The fork channel. Note that th e second term on the right -hand sid e dep end s on C whereas the remaining term s dep en d on C 2 . T aking C = Υ − 2 n C 0 ( γ ) w ith C 0 ( γ ) > 0 s mall enough and c = c 0 ( γ ) > 0 la rge enough, using ( 9 ) with the choi ce ε = c s Υ 2 n log b k r k r , w e get that P [ K a ≥ k r M t + c q Υ 2 n b k r log b k r ] ≤ P [ K a ≥ k r M t + k r ε ] ≤ 1 − O (log b k r ) k r k r ≤ b k − γ r . Note that ou r c hoice of ε satisfies Υ n C ε < 1 for k r a large enough p olynomial of n (co mpared to the sm all p olynomial Υ n ). A s im ilar inequalit y holds for the other directio n. Corr elate d sites. No w let T b e the f ork channel consisting o f no des r , u , a and b as in Figure 1 . Assume that a and b are con temp orary , call t the time separating them from u and d enote b y S ab the num b er of sites in a and b that are join tly sur viving from u . These are the sites th at pr o duce correlation b et w een the sequences at a an d b . All ot her sites are essentia lly noise. W e b ound the large deviations of S ab . Lemma 3.4 (F ork c h annel: large dev ations of join tly sur viving sites). Condition on the s e quenc e length a t u b eing k u . Then, for al l γ > 0 and al l b k u ≥ k u = n β ′′′ with β ′′′ > 0 lar ge enough, with c onditional pr ob ability at le ast 1 − b k − γ u , S ab = k u D 2 t ± S n ( q b k u log b k u ) , wher e the smal l p olynomial factor in S n ( q b k u log b k u ) dep ends on γ as wel l. ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 17 Pr oof. Eac h site in u surviv es in a with probab ility D t . Th e same holds for b indep endently . The result then follo ws fr om Chernoff ’s b oun d. W e ha v e P [ S ab < k u D 2 t − c q Υ 2 n b k u log b k u ] ≤ P " S ab < k u D 2 t − c s Υ 2 n log b k u k u · k u D 2 t # ≤ exp( − c 2 Υ 2 n D 2 t log b k u ) ≤ b k − γ u for c = c ( γ ) > 0 large enough, wh ere w e used ( 7 ). The other direction is similar. 3.2. Se quenc e p artitioning. F rom Lemma 3.2 , it follo ws that the sites of the ro ot sequence (or of an in ternal sequ en ce) remain fairly close to their exp ected p osition at the lea v es. W e take adv anta ge of this fact b y dividing eac h sequence in to blocks of size asymptotically larger than the t yp ical dis- placemen t implied b y Lemma 3.2 . As a result, matc hin g blo c ks in different sequences share a significan t fraction of sites. Moreo v er, distinct blo cks are roughly indep end en t. W e estimate the ev olutionary distance b et w een t wo lea v es by comparing the site f requencies in matc hing b lo c ks. This requires some care as w e sho w next. Consider the fork channel. W e seek to estimate the ev olutionary d istance b D ( a, b ) b et wee n a and b (normalize d b y the sequence length at u ). Partitioning the le af se quenc es. Let k 0 b e some deterministic length (to b e determined), and consider the fi rst k 0 sites in the sequences σ a and σ b at the n o des a and b , resp ectiv ely . If th e sequence at a or b has length smaller than k 0 , we decl are that our distance estimate e D ( a, b ) (see b elo w) is + ∞ . W e d ivide the leaf sequ ences into L blo cks of lengt h ℓ where ℓ = ⌈ k ζ 0 ⌉ , for some 1 2 < ζ < 1 to b e determined lat er and L = ⌊ k 0 /ℓ ⌋ . W e let k ′ 0 = ℓL . F or all i = 1 , . . . , L , we d efine th e i th blo ck σ a,i of a to b e the sub s equence of σ a ranging from p ositio n ( i − 1) ℓ + 1 to p osition iℓ . W e let Z a,i b e the n um b er of zeros ins ide σ a,i and define th e blo ck deviations ∆ a,i = Z a,i − ℓ 2 for al l i = 1 , . . . , L , and similarly for the sequ ence at b . Using the a b ov e notation w e defin e our distance estimator next. Assume that L is eve n. O therwise, w e can just drop the last b lo c k in the ab ov e partition. Our esti mator is e D ( a, b ) = 2 L L/ 2 − 1 X j =0 ∆ a, 2 j + 1 ∆ b, 2 j +1 . 18 C. DASKALAKIS AN D S. ROCH Notice that in our summation ab o v e w e skipp ed every other blo c k in our sequence partition to a v oid o verlapping sites and hen ce, decrease p otenti al correlations b et w een the terms in the estimator. In the rest of th is section, w e analyze the prop erties of e D ( a, b ) . T o do this it is helpful to consid er the sequence at u and the ev en ts that hap p ened in the c hannels defin ed b y the edges ( u, a ) and ( u, b ). Partitioning the anc estr al se q uenc e. Let u s c ho ose ℓ u to b e the largest in teger satisfying ℓ u M t ≤ ℓ. (10) Supp ose that the sequence σ u at nod e u is not shorter th an k ′ u = ( L − 1) ℓ u , and define the i th anc estr al blo ck σ u,i of u to b e the su bsequence of σ u ranging from p osition ( i − 1) ℓ u + 1 to p osition iℓ u , for all i ≤ L − 1. Giv en Lemma 3.2 , the c h oice of ℓ u in ( 10 ) is su c h that the blo cks of u and the corresp ondin g b lo c ks at a and b r oughly ali gn. In order to use the exp ecte d ev olutionary distance as computed in Lem- ma 2.4 , we define an “in terior” ancestral b lo c k whic h is guaran teed with high probabilit y to remain en tirely “inside” the corresp onding leaf blo c k. Let δ u = ⌈ L + 1 M t S n ( p k ′ u log k ′ u ) ⌉ , where the small p olynomial factor is the maxim um of th ose in the pro ofs of Lemma 3.2 and Lemma 3.4 for a giv en c h oice of γ . [The L = o ( √ k 0 ) in δ u is n eeded only when ( 10 ) is a strict inequalit y . S ee the pro of of Lemma 3.5 b elo w .] W e define the i th ( anc estr al ) interior blo ck σ ′ u,i of u to b e the sub s equence of σ u,i ranging from p osition ( i − 1) ℓ u + δ u of σ u to p osition iℓ u − δ u . Notice that δ u = S n ( √ k 0 log k 0 ), while ℓ u = S n ( k ζ 0 ). Therefore, for k 0 > k ∗ 0 , wh ere k ∗ 0 is su fficien tly large, ( i − 1) ℓ u + δ u < iℓ u − δ u so that the sequence σ ′ u,i is w ell defined. Also, for all i = 1 , . . . , L − 1, w e define x ′ a,i , y ′ a,i to b e th e p osition of the left-most (resp ., right -most) site in the sequence σ a descending fr om the site at p osition ( i − 1) ℓ u + δ u (resp., iℓ u − δ u of σ u ). Similarly , w e define x ′ b,i and y ′ b,i . Given this notation, we define the f ollo wing “go o d” ev ent E ′ 1 = {∀ i ≤ L − 1 : ( i − 1) ℓ < x ′ a,i , x ′ b,i < ( i − 1) ℓ + 2 M t δ u , (11) iℓ − 2 M t δ u < y ′ a,i , y ′ b,i < iℓ } . In tuitiv ely , when the ev ent E ′ 1 holds, all surviving descendan ts of the in terior blo c k σ ′ u,i are lo cated inside the blo c ks σ a,i and σ b,i, , resp ectiv ely (and the blo c ks remain large enough). T o argue ab out b lo c k in d ep end ence, we also defin e th e exterior blo ck σ ′′ u,i of u to b e the s u bsequence of σ u,i ranging from p osition ( i − 1) ℓ u − δ u of σ u to p osition iℓ u + δ u with co rresp onding p ositio ns x ′′ a,i , y ′′ a,i , x ′′ b,i and y ′′ b,i and ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 19 Fig. 2. Under the event E 1 the desc endants of the interior blo cks of σ u fal l inside the c orr esp onding blo cks of σ a ; the desc endants of the exterior blo cks of σ u c ontain al l surviving sites inside the c orr esp onding blo cks of σ a ; the windows of unc ertainty have l ength 2 M t δ u . go o d ev en t E ′′ 1 defined similarly as ab ov e, that is, E ′′ 1 = {∀ i ≤ L − 1 : ( i − 1) ℓ − 2 M t δ u < x ′′ a,i , x ′′ b,i < ( i − 1) ℓ, iℓ < y ′′ a,i , y ′′ b,i < iℓ + 2 M t δ u } . W e defin e E 1 = E ′ 1 ∪ E ′′ 1 . W e sh o w that th is ev en t holds with high p r obabilit y , conditioned on th e sequence length K u at u b eing at least k ′ u . Figure 2 sho ws the structure of the indel p ro cess in the case that the ev ent E 1 holds. Lemma 3.5 (In terior/exterior blo c k is inside/outside leaf blo ck). Con- ditione d on the event { K u ≥ k ′ u } , we have P [ E 1 ] ≥ 1 − 16 L 1 k ′ u γ . Pr oof. It follo ws f r om L emma 3.2 that the left-most d escendan t of the site at p osition ( i − 1) ℓ u + δ u of σ u is located inside the sequ ence of no d e a at p osition at least M t (( i − 1) ℓ u + δ u ) − S n ( p k ′ u log k ′ u ) > M t (( i − 1) ℓ u + L ) > ( i − 1) ℓ 20 C. DASKALAKIS AN D S. ROCH with probabilit y ≥ 1 − ( 1 k ′ u ) γ . The other b ounds follo w similarly . T aking a union b ound ov er all i ’s establishes the result. Blo ck c orr elation. Let S ab,i b e the n u m b er of common sites in the blo c ks σ a,i and σ b,i that are join tly surviving fr om u . Sim ilarly , w e define S ′ ab,i and S ′′ ab,i where, for ξ = a, b , σ ′ ξ ,i (resp., σ ′′ ξ ,i ) denotes the subsequence of σ ξ ranging f r om p osition x ′ ξ ,i (resp., x ′′ ξ ,i ) to p osition y ′ ξ ,i (resp., y ′′ ξ ,i ). W e define a goo d ev ent for S ab,i as E 2 = {∀ i ≤ L − 1 : ℓ u D 2 t − 3 M t δ u ≤ S ab,i ≤ ℓ u D 2 t + 3 M t δ u } . Lemma 3.6 (Join tly su rviving sites in blo cks). Conditione d on the event { K u ≥ k ′ u } , we have P [ E 2 ] ≥ 1 − 18 L 1 k ′ u γ . Pr oof. W e b ound P [ E c 2 ] = P [ E c 2 ∩ E 1 ] + P [ E c 2 ∩ E c 1 ] ≤ P [ E c 2 ∩ E 1 ] + P [ E c 1 ] ≤ P [ E c 2 ∩ E 1 ] + 16 L 1 k ′ u γ . By construction, u nder E 1 w e ha v e S ′ ab,i ≤ S ab,i ≤ S ′′ ab,i so th at P [ E c 2 ∩ E 1 ] ≤ P [ ∃ i, S ′ ab,i ≤ ℓ u D 2 t − 3 M t δ u ] + P [ ∃ i, S ′′ ab,i ≥ ℓ u D 2 t + 3 M t δ u ] ≤ P [ ∃ i, S ′ ab,i ≤ ( ℓ u − 2 δ u + 1) D 2 t − S n ( p k ′ u log k ′ u )] + P [ ∃ i, S ′′ ab,i ≥ ( ℓ u + 2 δ u + 1) D 2 t + S n ( p k ′ u log k ′ u )] ≤ 2 L 1 k ′ u γ b y Lemma 3.4 , where we also used the fact that D 2 t ≤ M t . 3.3. Estimation guar ante es. W e are no w ready to analyze the b ehavior of our estimat e e D ( a, b ). In this subsection w e compu te the exp ectation and v ariance of e D ( a, b ) . W e denote b y I a realization of the ind el p ro cess (b ut not of the substitution pro cess) on the paths b et w een u and a, b . W e denote b y E the ev ent th at { K u ≥ k ′ u } , E 1 and E 2 are satisfied. Supp ose that k 0 > k ∗ 0 (defined in S ection 3.2 ). ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 21 Lemma 3.7 (Block ind ep endence). Conditioning on I and E , the variables { ∆ a, 2 j + 1 ∆ b, 2 j +1 } L/ 2 − 1 j =1 ar e mutual ly indep endent. Pr oof. Observe that wh en K u ≥ k ′ u the ancestral blo c ks σ u,i are well defined. Assuming that k 0 > k ∗ 0 , the interior b lo c ks σ ′ u,i are also well defin ed and d isjoin t. Hence, for a fixed I under E , f or all i ≤ L − 1, b oth ∆ a,i and ∆ b,i dep end on the su bsequence of σ u ranging from p osition ( i − 1) ℓ u − δ u + 1 to p ositio n iℓ u + δ u − 1. In this case, for j ∈ { 1 , . . . , L / 2 − 1 } , differ- en t ∆ a, 2 j + 1 ∆ b, 2 j +1 ’s are fu nctions of differen t su bsequences of σ u . Obs erv e that, since the ro ot sequence is i.i.d. u niform and the in s ertions ab o ve u are also i.i.d. uniform, the state of ev ery site in σ u is un if orm and inde- p end ent from the other sites. It f ollo ws from the ab ov e observ ations that { ∆ a, 2 j + 1 ∆ b, 2 j +1 } L/ 2 − 1 j =1 are mutually indep endent. Lemma 3.8 (Exp ected correlation under goo d ev ent). We have E [∆ a,i ∆ b,i |I , E ] = 1 4 e − 4 ηt e − 2 µt ℓ u ± S n ( p k 0 log k 0 ) . Pr oof. Let ∆ S a,i b e the contribution to ∆ a,i from th ose common sites b et w een a an d b th at are jointly surviving from u . Let ∆ NS a,i = ∆ a,i − ∆ S a,i , and similarly f or b . Then E [∆ a,i ∆ b,i |I , E ] = E [(∆ S a,i + ∆ NS a,i )(∆ S b,i + ∆ NS b,i ) |I , E ] = E [∆ S a,i ∆ S b,i |I , E ] , since the con tribu tion fr om ∆ NS a,i and ∆ NS b,i is in dep end en t and a v erages to 0. W rite ∆ S a,i as a sum o ve r the join tly surviving sites, that is, ∆ S a,i = S ab,i X j =1 z ( j ) a,i − 1 2 , where z ( j ) a,i is 1 if the corresp onding site of a is 0. Note that the terms in paren theses ha v e zero e xp ectation giv en I and E . Then, E [∆ S a,i ∆ S b,i |I , E ] = S ab,i X j =1 E z ( j ) a,i − 1 2 z ( j ) b,i − 1 2 I , E b y ind ep endence of the sites. W e compute the exp ectation ab o ve. W e ha v e E z ( j ) a,i − 1 2 z ( j ) b,i − 1 2 I , E = E z ( j ) a,i z ( j ) b,i − 1 2 z ( j ) a,i − 1 2 z ( j ) b,i + 1 4 I , E 22 C. DASKALAKIS AN D S. ROCH = E [ z ( j ) a,i z ( j ) b,i |I , E ] − 1 4 = 1 2 1 + e − 4 ηt 2 − 1 4 = 1 4 e − 4 ηt . Therefore, E [∆ S a,i ∆ S b,i |I , E ] = 1 4 e − 4 ηt S ab,i . The result then follo ws fr om the definition o f E 2 . Lemma 3.9 (V ariance under goo d ev ent) . We have V ar[∆ a,i ∆ b,i |I , E ] ≤ 3 16 ℓ 2 . Pr oof. By Cauc hy–Sc h warz w e ha ve E [∆ 2 a,i ∆ 2 b,i |I , E ] ≤ ( E [∆ 4 a,i |I , E ] E [∆ 4 b,i |I , E ]) 1 / 2 = ( 1 16 (3 ℓ 2 − 2 ℓ ) · 1 16 (3 ℓ 2 − 2 ℓ )) 1 / 2 ≤ 3 16 ℓ 2 , where we u s ed the fact th at the length of the sequences σ a,i and σ b,i is deterministically ℓ , and th e num b er of zeros in σ a,i and σ b,i follo ws a b inomial distribution with ℓ trials and probabilit y 1 / 2. Lemma 3.10 (Distance estimate). We have E [ e D ( a, b ) |I , E ] = 1 4 e − (4 η + µ + λ ) t ℓ ± S n ( p k 0 log k 0 ) and V ar [ e D ( a, b ) |I , E ] ≤ 3 8 1 ⌊ k 1 − ζ 0 ⌋ ℓ 2 . In p articular, the standar d deviation STD[ e D ( a, b ) |I , E ] = O ( k (3 ζ − 1) / 2 0 ) = o ( p k 0 ) for ζ > 1 / 2 smal l enough. Pr oof. F r om Lemma 3.7 , th e L/ 2 = ⌊ k 0 /ℓ ⌋ / 2 terms in e D ( a, b ) are mu- tually indep endent. The pro of then f ollo ws from Lemmas 3.8 and 3.9 and the definition of ℓ u . ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 23 3.4. Conc e ntr ation. W e no w sho w that our distance esti mate is concen- trated. F or notational con venience, we d enote b y P ∗ u the p robabilit y measure induced b y conditioning on the ev en t { K u ≥ k ′ u } . Reca ll th at the ev ent E is con tained in { K u ≥ k ′ u } . Lemma 3.11 (Concen tration of distance estimate) . L et α > 0 b e such that ζ − α > 1 / 2 , and β = 1 − ζ − 2 α > 0 for ζ > 1 / 2 smal l enough. Then for k 0 lar ge enough P ∗ u 4 ℓ e D ( a, b ) − e − (4 η + µ + λ ) t > 1 k α 0 ≤ O 1 k β 0 . Pr oof. W e us e C heb yshev’s inequ alit y . W e first co ndition on I , E . Re- calling that ℓ = ⌈ k ζ 0 ⌉ , note that P ∗ u 4 ℓ e D ( a, b ) > e − (4 η + µ + λ ) t + 1 k α 0 I , E ≤ P ∗ u e D ( a, b ) > ℓ 4 e − (4 η + µ + λ ) t + ℓ 4 1 k α 0 I , E ≤ P ∗ u e D ( a, b ) > E [ e D ( a, b ) |I , E ] − S n ( p k 0 log k 0 ) + ℓ 4 1 k α 0 I , E ≤ 3 ℓ 2 / (8 ⌊ k 1 − ζ 0 ⌋ ) ( ℓ/ (4 k α 0 ) − S n ( √ k 0 log k 0 )) 2 = O 1 k 1 − ζ − 2 α 0 . The other d irection is similar. T aking exp ectation o ve r I , we ha v e P ∗ u 4 ℓ e D ( a, b ) − e − (4 η + µ + λ ) t > 1 k α 0 E ≤ O 1 k β 0 . Cho ose γ > 0 in Lemmas 3.2 an d 3.4 large enough so that γ − (1 − ζ ) > β . Then, from Lemm as 3.5 and 3.6 , w e ha v e P ∗ u 4 ℓ e D ( a, b ) − e − (4 η + µ + λ ) t > 1 k α 0 ≤ P ∗ u 4 ℓ e D ( a, b ) − e − (4 η + µ + λ ) t > 1 k α 0 E P ∗ u [ E ] + P ∗ u [ E c ] ≤ O 1 k β 0 . The pro ofs of Theorems 1 and 2 are g iv en in the next section. 24 C. DASKALAKIS AN D S. ROCH 4. Putting it all toget her. L ar ge-sc ale asymptotics. W e are ready to pr o ve our main result in the molecular cloc k case. W e p ostp one the more general case to the next section. A last b it of notation: for a p air of lea ve s a, b ∈ [ n ], w e d enote b y t ab the time b et wee n a , b and their m ost recen t common a ncestor. Pr oof of Theore m 2 . W e first giv e a b ound on the diameter of the tree. Let h (resp., H ) b e the length of the s hortest (r esp ., longest) p ath b et w een the ro ot and a leaf in graph distance. Because the n um b er of leav es is n w e m ust ha ve 2 h ≤ n and 2 H ≥ n . Since all lea v es are contemporaneous it m u st b e that H f ≤ hg . Com b ining these constrain ts giv es that the diameter Diam satisfies 2 f g log 2 n ≤ 2 h ≤ Dia m ≤ 2 H ≤ 2 g f log 2 n. Giv en our b ound on the diameter of the tree, it follo ws that the time from the ro ot r of the tree to any leaf is at most g 2 f log 2 n . Supp ose that the length k r at the ro ot of the tree satisfies k r > k ∗ r = k ∗ r ( k 0 ), wh ere k ∗ r is the minim u m in teger satisfying k ∗ r ≥ e ( g 2 /f ) · µ l og 2 n ( k 0 + S n ( p k ∗ r log k ∗ r )) , where the small p olynomial factor is tak en to b e the one used in Lemma 3.2 . Lemma 3.2 and a union b ound then imply that w ith probabilit y at le ast 1 − O ( n ) · ( k ∗ r ) − γ for al l no des u K u ≥ k ′ u . Lemma 4.1 (Concen tration of distance estimate). F or al l α ′ > 0 , β ′ > 0 , ther e exists k 0 = n β ′′′ with β ′′′ > 0 lar ge enough so tha t if the se quenc e length at the r o ot is k r > k ∗ r ( k 0 ) , then P ∀ a, b ∈ [ n ] , 4 ℓ e D ( a, b ) − e − (4 η + µ + λ ) t ab ≤ 1 n α ′ = 1 − O 1 n β ′ . Pr oof. This follo ws from Lemma 3.11 and our observ ation ab o ve th at, if k r > k ∗ r ( k 0 ), with probabilit y at least 1 − O ( n ) · ( k ∗ r ) − γ , then K u ≥ k ′ u for all no des u . Giv en our b oun d on the diameter of the tree, it follo w s that for all pairs of lea v es a , b and sm all ε > 0 e − (4 η + µ + λ ) t ab ± ε = e − (4 η + µ + λ ) t ab (1 ± O ( ε )) ≥ 1 n α ′′ (1 ± O ( ε )) . ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 25 Therefore, c h o osing α ′ large enough in L emm a 4.1 , w e get that all distances can b e estimated within a s mall ε sim u ltaneously with p robabilit y going to 1. Using the standard Bun eman algorithm, we can reco ver the tree efficiently (see, e.g., [ 34 ]). Constant-size c ase. The pr o of of T heorem 1 for the molecular clo ck case builds on the pro of of Theorem 2 by tr eating n as a constant and letting the sequence length a t the r o ot o f the tree go to in finit y . Pr oof of Theor e m 1 (Molecular clo ck case) . W e can restate Lem- ma 4.1 in the follo wing f orm, where the failure probabilit y is expressed more cleanly in terms of the sequence length at the ro ot o f the tree. The p ro of of the lemma is essen tially th e same. Lemma 4.2 (Concent ration of distance estimate). F or al l α ′ > 0 , ther e exists k ∗ 0 = n β ′′′ for β ′′′ > 0 lar ge enough such that if the se quenc e length at the r o ot is k r > k ∗ r ( k ∗ 0 ) , then P ∀ a, b ∈ [ n ] , 4 ℓ e D ( a, b ) − e − (4 η + µ + λ ) t ab ≤ 1 n α ′ = 1 − O ( n · k − γ r ) − O ( n 2 · k − β r ) . Rep eating the pro of of Theorem 2 ab o v e, it follo ws that the algorithm fails to reconstruct the phylog en y with probabilit y O ( n · k − γ r ) + O ( n 2 · k − β r ). Letting k r → + ∞ co n cludes the pro of of Theorem 1 . 5. Extensions. GTR mo del. W e briefly discuss h o w our results can b e extended to GTR mo dels. F or bac kground on GTR mo dels, see, for example, [ 16 ]. Let Q b e a rev ersible 4 × 4 rate matrix with stationary distribution π . Our new se- quence ev olution pro cess is identica l to the one d escrib ed in Definition 1.1 except that the su bstitution pro cess is a con tinuous-time Mark ov pro cess with rate matrix η e Q . The rate matrix Q has 4 nonn egativ e eigen v alues. F or con venience, we assume that the largest negativ e eigen v alue is − 1. W e denote by w the corresp on d ing eigen v ector whic h w e assume is normalized as X s ∈{ A , G , C , T } π s w 2 s = 1 . W e no w p erform the f ollo wing transformation of the state s p ace. F or a no de u , let σ u = ( σ 1 u , . . . , σ K u u ) b e the transformed sequence at u w h ere 26 C. DASKALAKIS AN D S. ROCH σ i u = w A (resp., w G , w C , w T ) if the state at site i is A (resp., G , C , T ). Note that, under stationarit y , the exp ectation of the state at site i is 0 by orthog- onalit y of π and w . Then our d istance esti mator is b D ( a, b ) = K a X i =1 σ i a ! K b X j =1 σ j b ! . In p articular, in the t w o-state CFN case, we h a ve w = (+1 , − 1) and w e obtain th e s ame estimate as b efore, u p to a constant. W e no w analyze the exp ectation of this quantit y . F or u ∈ V , w e let ∆ u = K u X i =1 σ i u . Lemma 5.1. The fol lowing holds: E [∆ a | σ r ] = e − ( η + µ ) t ∆ r . (12) Remark 5.2. Note that th is formula is sligh tly differen t than that in Lemma 2.2 b ecause of the norm alization im p lied by requiring Q to ha ve second eigen v alue − 1. Pr oof of Lemma 5.1 . The sites created after r con tribu te 0 in exp ec- tation. Of course, so d o the deleted sites. The fraction of sites that surviv e is e − µt . S upp ose site i surviv es, then note that E [ σ i a | σ i r = w s , i survive s] = X s ′ ( e ηtQ ) ss ′ w s ′ = e − ηt w s . Summing o v er all sites of r w e get E [∆ a | σ r ] = e − ( η + µ ) t ∆ r as clai med. Consider n o w a “fork” tr ee, that is, a ro ot r from whic h emanates a single edge e u = ( r , u ) whic h in turn branc hes in to t wo edges e a = ( u, a ) and e b = ( u, b ). F or x = a, b, u , w e denote the parameters of edge e x b y t x , λ x , µ x , η x . Our goal is to compu te E [ b D ( a, b )] assu m ing that the sequence length at the ro ot is k . The p ro of is similar to Lemma 2.3 . Lemma 5.3. The fol lowing holds: E [ b D ( a, b )] = e − ( η a + µ a ) t a e − ( η b + µ b ) t b e − ( µ u − λ u ) t u k . Note that R emark 5.2 also applies her e. ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 27 Pr oof. W e hav e E [ b D ( a, b )] = E [∆ a ∆ b ] = E [ E [∆ a ∆ b | σ u ]] = E [ E [∆ a | σ u ] E [∆ b | σ u ]] = e − η a t a e − µ a t a e − η b t b e − µ b t b E [∆ 2 u ] = e − η a t a e − µ a t a e − η b t b e − µ b t b E [ E [∆ 2 u | K u ]] = e − η a t a e − µ a t a e − η b t b e − µ b t b E [V ar[∆ u | K u ]] = e − η a t a e − µ a t a e − η b t b e − µ b t b E [ K u V ar[ σ 1 u ]] = e − η a t a e − µ a t a e − η b t b e − µ b t b E [ K u E [( σ 1 u ) 2 ]] = e − η a t a e − µ a t a e − η b t b e − µ b t b e − ( µ u − λ u ) t u k b y Lemma 5.1 . F rom the pr evious lemmas, on e can adapt the pro ofs ab o v e to the GTR case. Nonclo ck c ase. Using Lemma 5.3 , w e can get rid of th e molecula r clo ck assumption. Cons id er again the fork tree, but assume that eac h edge is in fact a path. An adaptatio n of Lemma 5.3 giv es the foll o wing lemma. Lemma 5.4. The fol lowing holds: − ln E [ b D ( a, b )] p E [ K a ] E [ K b ] = X e ∈ P( a,b ) ( η e + µ e / 2 + λ e / 2) t e . Note that R emark 5.2 also applies her e. Pr oof. Note that − ln ( k − 1 E [ K a ]) = X e ∈ P( r,a ) ( µ e − λ e ) t e and similarly f or b . A v arian t of Lemm a 5.3 giv es − ln( k − 1 E [ b D ( a, b )]) = X e ∈ P( a,b ) ( η e + µ e ) t e + X e ∈ P( r,u ) ( µ e − λ e ) t e . The result follo w s by subtracting the p revious expressions. The expression in Lemma 5.4 provides the additiv e metric needed to ex- tend our resu lts to non clo ck b ounded-rates case. 28 C. DASKALAKIS AN D S. ROCH 6. Concluding remarks. W e ha ve sho wn ho w to reco n struct p h ylogenies under the b ounded-r ates, GTR mo del with indels. Ou r efficient algorithm requires p olynomial-lengt h sequences at the ro ot. A n atural op en pr oblem arises f r om this work: Can our results b e extended to general trees w ith b ound ed branc h lengths, as opp osed to the b oun ded-rates mo del? The key difference b et wee n the t wo m o dels is that the former ma y ha v e a linear diameter whereas the latter has logarithmic diameter. T o extend our r esults, one w ould need to d eal with far a w ay lea ve s that are almost un correlated but for wh ic h our block stru cture does not apply . REFERENCES [1] Andoni, A. , Bra verma n, M. and Hassidim, A. (2011). Phylogenetic reconstruction with insertions and deletions. Preprint. [2] Andoni, A. , Daskalakis, C . , Hassid im, A. and R och, S. (2010). Global alignment of molecular sequences v ia ancestral state reconstruction. I n ICS 2010 358–36 9. Tsingh u a U niversit y Press, Beijing, China. [3] A threy a, K. B. and Ney, P. E. (1972). Br anching Pr o c esses . Die Grund lehr en der Mathematischen Wissenschaften 196 . Sp ringer, New Y ork. MR0373040 [4] A tteson, K. (1999). The p erformance of neighbor-joining metho ds of phylogenetic reconstruction. Algorithmic a 25 251–278. MR1703580 [5] Csur ¨ os, M. (2002). F ast recov ery of evolutionary trees with t h ousands of no des. J. Comput. Biol. 9 277–297. [6] Csur ¨ os, M. and Kao, M.-Y . (2001). Prov ably fast and accurate recov ery of evolu- tionary trees through harmonic greedy triplets. SIAM J. Comput. 31 306–322 (electronic). MR1857402 [7] D askalakis, C. , Hi ll, C. , Jaffe, A. , Miha e scu, R. , Mossel, E. and Rao, S. (2006). Maximal accurate forests from distance matrices. In RECOMB 2006 281–295 . Springer, Berlin. [8] D askalakis, C. , Mossel, E. and Roch, S. (2006). Optimal phylogenetic recon- struction. In STOC’06: Pr o c e e dings of the 38th Annual ACM Symp osium on The ory of Computing 159–168. A CM, New Y ork . MR2277141 [9] D askalakis, C. , Mossel, E. and Ro ch, S . ( 2009). Ph y logenies without branch b ounds: Contracting the short, pruning the deep. I n RECOMB 2009 451–465 . Springer, Berlin. [10] Daskalak is, C. and Roch, S. (2010). Alignment-free phylogenetic reconstru ct ion. In RECOMB 2010 123–137. S pringer, Berlin. [11] Edgar, R. C. (2004). MUSCLE: Multiple se quence alignmen t with high accuracy and high through p ut. Nucleic A cids R es. 32 1792–17 97. [12] Elias, I. (2006). Settling the intractabili ty of multiple alignmen t. J. Comput. Bi ol. 13 1323–1339 ( electronic). MR2264995 [13] Erd ˝ os, P. L. , Steel, M. A. , Sz ´ ekel y , L. A. and W arnow , T. J. (1999). A few logs suffice to build (almost) all trees. I. R andom Structur es A l gorithms 14 153–1 84. MR1667319 [14] Erd ˝ os, P. L. , S teel, M. A. , Sz ´ ekel y , L. A. and W arnow , T. J. (1999). A few logs suffice to build (almost) all trees. I I. The or et. Com put. Sci. 221 77–118 . MR1700821 [15] Felsenstein, J. (1978). Cases in whic h p arsimon y or compatibilit y meth od s will b e p ositiv ely misleading. Syst. Bi ol. 27 401–410. ALIGNMENT-FREE PH Y LOGENETIC RECONSTRUCTION 29 [16] Felsenstein, J. (2004). Inferring Phylo genies . S inauer, New Y ork. [17] Graur, D. and Li, W. H. (1999). F undamentals of Mole cular Evolution , 2nd ed. Sinauer, Sun derland, MA. [18] Grona u, I. , Moran, S. and Snir, S. (2008). F ast and reliable reconstruction of phylogenetic trees with very short edges (extended abstract). In Pr o c e e dings of the Ni nete enth Annual ACM-SIAM Symp osium on Di scr ete Algorithms 379–388. ACM , New Y ork. MR2487605 [19] Higgins, D. G. and Sharp, P. M. (1988). Clustal: A p ac k age for p erformi ng multiple sequence alignment on a micro computer. Gene 73 237–244 . [20] Huson, D. H. , Nettles, S. H . and W arn o w, T. J. (1999). Disk-co vering, a fast- conv erging metho d for phylogenetic tree reconstruction. J. Comput. Biol. 6 3–4. [21] Karlin, S. and T a ylor, H. M. (1981). A Se c ond Course in Sto chastic Pr o c esses . Academic Press, New Y ork. MR0611513 [22] Ka toh, K. , Misa w a, K. , Kuma, K.-i. and Miy a t a, T. (2002). MAFFT: A nov el metho d for rapid multiple sequ ence alignmen t b ased on fast F ourier transform. Nucleic A cids R es. 30 3059–3 066. [23] King, V . , Zhang, L. and Zhou, Y. (2003). On th e complexity of distance-based evol utionary tree reconstruction. In Pr o c e e dings of the F ourte enth Annual ACM- SIAM Symp osium on Discr ete Algorithms (Baltim or e, M D, 2003) 444–453. ACM , New Y ork. MR1974948 [24] Lacey, M. R. and Chang, J. T. (2006). A signal-to-noise analysis of phylogeny estimation by neighbor- joining: Insufficiency of p olynomial length sequences. Math. Biosci. 199 188–215. MR2211625 [25] Liu, K. , Ra g h a v an, S. , Nelesen, S. , Linder, C. R. and W arnow, T. (2009). Rapid and accurate large-scale co estimation of sequ ence alignments and phylogenetic trees. Scienc e 324 1561–1 564. [26] L ¨ oytynoja, A. and Goldman, N. (2008). Phylogen y- aw are gap placement preven ts errors in seq u ence alignmen t and ev olutionary analysis. Scienc e 320 1632–1 635. [27] Metzler, D. (2003). Statistical alignmen t b ased on fragmen t insertion and d eletion mod els. Bioinformatics 19 490–499. [28] Miklos, I. , Lunter, G. A. and Holmes, I. (2004). A “Long I ndel” mo del for evol utionary sequence alignment. Mol. Biol. Evol. 21 529–540. [29] Mossel, E. (2007). Distorted metrics on trees and phylogenetic forests. IEEE/ACM T r ans. Comput. Bi o. Bioinform. 4 108–116. [30] Mossel, E. and Roch, S. (2006). Learning nonsingular p hylogenies and hidden Mark o v mo dels. Ann. Appl. Pr ob ab. 16 583–614. MR2244426 [31] Motw a n i, R. and Ra gha v an, P. (1995). R andomi ze d Algorithms . Cam b ridge Univ. Press, Cam bridge. MR1344451 [32] Riv as, E. and Eddy, S. R . (2008). Probabilistic p hylogenetic inference with inser- tions and d eletions. PL oS Comput. Biol. 4 e10001 72, 20. MR2448496 [33] Roch, S. (2008). Sequence-length requ irement for distance-based phylogen y recon- struction: Breaking the polyn omial b arrier. In FOCS 2008 729–738. IEEE Com- put. So c., Los A lamitos, CA. [34] Semple, C. and Stee l, M. (2003). Phylo genetics . Oxfor d L e ctur e Series in Mathe- matics and Its Applic ations 24 . Ox ford Univ. Press, O x ford. MR2060009 [35] Steel, M. A. and Sz ´ ekel y , L. A. (1999). Inv erting random funct ions. A nn. Comb. 3 103–113. MR1769697 [36] Steel, M. A . an d Sz ´ ekel y , L. A. (2002). Inv ertin g random functions. I I. Exp licit b ounds for d iscrete maximum likelihood estimation, with applications. SIAM J. Discr ete Math. 15 562–575 (electronic). MR1935839 30 C. DASKALAKIS AN D S. ROCH [37] Suchard, M. A. and Rede lings, B . D. (2006). BAli-Ph y: S im ultaneous Ba yesian inference of alignment and phylogeny. Bioinf ormatics 22 2047–204 8. [38] Tha tte, B. D. (2006). Invertibilit y of the TKF mo del of sequence evolutio n. Math. Biosci. 200 58–75. MR2211928 [39] Thorne, J. L. , Kishino, H. and Felsenstein, J. (1991). An evolutionary mo del for maximum likelihoo d alignment of d na sequences. Journal of Mol e cular Evolution 33 114–124. [40] Thorne, J. L. , Kishi n o, H. and Felsenstein, J. (1992). I nching to ward realit y: An impro ved likelihood mod el of sequence ev olution. Journal of M ole cular Evolution 34 3–16. [41] W ang, L. and Jiang, T. (1994). On the complexity of multiple sequence alignmen t. J. Comput. Biol. 1 337–348. [42] Wong, K. M. , S u chard, M. A. and Huelsenbeck, J. P. (20 08). Alignmen t un- certain ty and genomic analysis. Scienc e 319 473–476. MR2381044 Dep ar tment of Electrical Engineering and Computer Science Massachusetts Institute of Technology 32 V a ssar Street Cambridge, Massachusetts 02139 USA E-mail: costis@csail.mit.edu Dep ar tment of Ma them atic s and Bioinforma tics Program University of California, Los Ang eles 520 Por tola Plaza Los Angeles, California 90 095-155 5 USA E-mail: ro c h@math.ucla.edu
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment